Giant cell arteritis (GCA) is a commonly occurring large vacuities characterized by angiopathy of medium and large-sized vessels. GCA granulomatous formation plays an important role in the pathogenesis of GCA. Analysis of T cell lineages and signaling pathways in GCA have revealed the essential role of T cells in the pathology of GCA. T cells are the dominant population present in GCA lesions. CD4+ T cell subtypes that are present include Th1, Th2, Th9, Th17, follicular helper T (Tfh) cells, and regulatory T (Treg) cells. CD8 T cells can primarily differentiate into cytotoxic CD8+ T lymphocytes and Treg cells. The instrumental part of GCA is the interplay between dendritic cells, macrophages and endothelial cells, which can result in the vascular injury and the characteristics granulomatous infiltrates formation. During the inflammatory loop of GCA, several signaling pathways have been reported to play an essential role in recruiting, activating and differentiating T cells, including T-cell receptor (TCR) signaling, vascular endothelial growth factor (VEGF)-Jagged-Notch signaling and the Janus kinase and signal transducer and activator of transcription (STAT) pathway (JAK-STAT) pathway. In this review, we have focused on the role of T cells and their potential signaling mechanism (s) that are involved in the pathogenesis of GCA. A better understanding of the role of T cells mediated complicated orchestration during the homeostasis and the changes could possibly favor developments of novel treatment strategies against immunological disorders associated with GCA.
Giant cell arteritis (GCA) is a chronic granulomatous vacuity of the large and middle-sized arteries, mainly affecting the thoracic aorta branches.[1-3] GCA mainly affects the women over 50 years. GCA prevalence has been reported to increase with age, and generally peak during the eighth decade of life.[4] GCA poses a significant threat to the patient’s life and adversely affects their quality of life. Without efficient treatments, GCA patients face devastating complications (including ocular and neurologic complications) and high mortality.[5] Currently, the therapy of GCA still depends on the application of glucocorticoids as the first-line treatment, which can lead to adverse effects, such as hypertension and hyperglycemia. However, no methods can cure giant cell arteritis until now due to the complicated pathophysiology. To date, the underlying pathogenesis of GCA remains elusive. A number of previous studies have postulated that the clonal expansion of T cells in the vascular lesions may play an important role in GCA. In addition, PLG and P4HA2 have been identified as key genetic factors that could increase the susceptibility to GCA in a genome-wide association studies genome-wide association studies (GWASs).[6] T-cells were also found to outnumber B-cells in the temporal artery biopsies (TABs) of GCA patients.[7] These reports provided a significant evidences that GCA primarily occurs as a result of T cells reaction to human leukocyte antigen (HLA) molecules. In this review, we have discussed the potential role of T cells and its subtypes, and analyzed the T cell signaling pathway that has been implicated in the pathogenesis of GCA.
MAJOR SUBGROUPS OF T CELLS AND THE CHARACTERISTICS AND FUNCTIONS
CD4+ T Cells in GCA
A thorough probing about the role of CD4+ T cells contributes to the understanding of the complete pathogenesis of GCA. In the healthy controls, CD4+ T cells do not populate in the arterial walls. However, in GCA, the enhanced accumulation of CD4+ T cells in the transmural infiltrates of GCA-affected arteries have confirmed their crucial role under both in vitro and in vivo settings.[8] More importantly, it has been reported that the reduced number of CD4+ T cells in the GCA artery in severe combined immune-deficient (SCID) murine models can significantly deplete the arteritis lesions. Firstly, CD4+ T cells are recruited in the adventitia via the various chemokine (such as CCL18, CCL19, CCL20 and CCL21) synthesized by the dendritic cells (DCs). In particular, CCL20 plays an essential role in triggering CD4+ T cells recruitment by binding to its specific ligand (CCR6). Once the T cells are recruited and activated, they can effectively differentiate into CD4+ T cells and CD8+ T cells. CD4+ T cells include effector CD4+ T cells and CD4+regulatory T cells. The subtypes of CD4+ T cells include Th1 cells, Th2 cells, Th9 cells, Th17 cells and Th21 cells. CD8+ T cells subsets include effector CD8+ T cells and CD8+FOXP3+ Treg cells. CD4+ T cells as well as CD8+ T cells and polarizing into specialized subsets need optimal cytokine milieu via celluar signaling pathways.[9]
Th1 cells are the most highly expressed among the all-differentiated T cells subpopulations in the circulation of the healthy individuals, as effector cells involved in the pathogenesis of giant cell arteritis.[10,11] GCA has been primarily envisioned as a Th1-induced disease due to the characteristic of the granulomatous in the giant cells.[12] The number of Th1 cells has been reported to be elevated in the peripheral blood and granulomatous infiltrates of GCA patients.[13] Recent studies have also revealed that Th1 cells lineage may be directly derived from CD4+ T cells in the presence of IL-12 and IL-18,[14,15] or from the local polarization of Th17 cells into Th1/Th17 cells in the presence of IL-12.[16] Differentiation of Th1 cells depends on the activation of Janus kinase (JAK) and signal transducer and activator of transcription (STAT) signaling (including STAT1, STAT4) cascades[17] that play a crucial role in the development of Th1 cells.[18] Th1 cells can also contribute to the generation of antibodies by B-lymphocytes, such as immunoglobulin G(IgG), immunoglobulin M(IgM), and immunoglobulin A(IgA) in the mouse model or the healthy subjects.[19] Interestingly, suppression of JAK-STAT signaling by Tofacitinib, a JAK1/3 inhibitor, could markedly decrease the percentage of Th1-lineage committed cells in vitro as well as the deregulated production of Interferon-gamma (IFN-γ) via the suppression of Th1 cells.[20] IFN-γ, as the signature cytokine of Th1 cells, is overexpressed in the serum and adventitial of newly patients with GCA,[21] thereby closely resembling the degree of intramural vessel formation in the affected temporal artery.[20,22]
Moreover, a strong suppressive effect of Tofacitinib on the microvessel angiogenesis has been reported, which resulted in the lesser amounts of micro vessels in SCID mice than those in the vehicle control arteries, thus highlighting the pivotal role of JAK1/3-dependent cytokine signaling in the development of GCA vasculopathy.[20]
Compared with Th1 cells, Th2 cells express different receptors, including chemokine (C-C motif) receptor 3(CCR3) (whose ligands is CCL11), CCR4 (whose ligands are CCL11, CCL22 and CCL17), and CCR8 (whose ligands is CCL11).[23] Mast cells can effectively promote CD4+ T cells proliferation and differentiation into Th2 cells through the communication with CD4+ T cells. Moreover, Th2 polarization can be regulated by STAT6, as the mRNA and protein expression of this transcription factor was found to be significantly elevated in the lesion vessels. However, Th2-derived cytokines were found to be absent in GCA, whereas IL-33, that can to stimulate Th2 production, both at the mRNA and protein level was overexpressed in temporal artery biopsies (TABs) of the inflamed arteritis of GCA patients with strong alternatively activated macrophage (M2) polarization.[24] Another important cytokine produced by Th2 cells is IL-9, which is associated with the presence of IL-33 or transforming growth factor β (TGF-β) separated from the peripheral blood.[25] Francesco et al, unveiled that IL-9 has an important regulatory role on the pro-inflammatory functions of the various immune cells in GCA, including antigen-presenting cells, Th17 cells and Treg cells.[26,27] IL-9 expression can also be detected in the affected vessels. IL-9 binds to IL-9R accompanied with the release of IL-8, which can correlate with the transmural inflammation and vasa vasorum vacuities.[28] Moreover, expression of IL-9 in giant cells of GCA, along with IL-17, can correlate with the strength of the systemic inflammatory response produced.[27] Nevertheless, the elucidation of other functions of Th2 cells in GCA pathogenesis requires further studies.
Additionally, IL-9-secreting-Th9 cells can abundantly accumulate in the GCA-affected-artery. Th9 cells differentiation requires the presence of stimulatory cytokines (IL-4 together with TGF-β).[27] IL-9 cannot be inhibited by glucocorticoids and might perpetuate the chronicity of tissue injury.[29] Thereby, targeting Th9 cells and IL-9 may be a promising strategy to eliminate chronic inflammation and thus reduce the intensity of GCA.
The increasing frequencies of Th17 cells can be found in both the vascular lesions and blood of untreated GCA patients in contrast with the healthy subjects.[10,11,16] Th17 polarization is regulated by various differentiating factors including IL-21, IL-23, IL-1β, IL-6 and TGF-β.[13,30] Th17 cells and Tfh cells can synthesize IL-21 whereas it can effectively promote the differentiation of Th17 cells in the absence of IL-6 and IL-23.[31] IL-23 mainly contributes to the maintenance and advanced maturation of Th17 cells. TGF-β together with IL-6 can initiate Th17 differentiation by inducing the expression of RORγt (a symbol transcription factor for Th17 cells encoded by Rorc (γt))) to alter the balance between Th17 cells and Foxp3+ regulatory T (Treg) cells.[31]
Th17 cells are characterized by the production of IL-17A, which can drive a robust chronic inflammatory immune response in many autoimmune diseases, and therapies targeting IL-17A has demonstrated significant efficacy in psoriasis.[32] An elevated IL-17A level has been detected in the local lesions of artery. In addition, IL-17A can play an important role in the repair of artery wall. The levels of IL-17 protein and mRNA have been observed to decrease rapidly after the glucocorticoid treatment.[33] Moreover, an increase in IL-17 expression in the temporal artery lesions indicates perpetual response to the glucocorticoid treatment and a low risk of relapse in patients with GCA.[34,35] IL-17A has a significant effect on the various types of somatic cells (such as fibroblasts, endothelial cells, and vascular smooth muscle cells) and can regulate the synthesis of multiple pro-inflammatory cytokines (including endothelial adhesion molecules, chemokine, metalloproteinase, IL-1β, IL-6 and TNFα).[36] In addition, Th17 cells can also stimulate the production of other important cytokines, such as IL-22 and IL-26. IL-22 can also maintain the homeostasis of epithelial cells. Thus, the role of IL-26 in pathogenesis of GCA needs to be investigated in detail.
Follicular helper T (Tfh) cells, the second most frequent tissue-resident T cells, account for 10% of the whole T cells populations.[37] Tfh cells can produce IL-21, which could be detected in the arterial lesions as well as the peripheral blood of GCA patients.[37] IL-21 can contribute to the differentiation of Tfh cells and Th17 cells, balance the helper T cells subtypes, enhance B cells differentiation into the plasma cells and promote the production of immunoglobulin via the stimulation of the germinal centers.[38-40]
Treg cells have been implicated in maintaining tolerance and preventing autoimmunity inflammation.[41] In patients with GCA, impaired function and decreased number of Treg cells in the peripheral blood as compared with healthy individuals can lead to the insufficient self-tolerance and autoimmunity inflammation.[10] Treg cells defined by the expression of FoxP3, as a transcription factor, that is primarily involved in the development of CD4+ Treg cells.[42] IL-6 and TGF-β together can trigger Th17 polarization but TGF-β alone leads to the differentiation of Treg cells.[43] Treg cells can also differentiate into three functionally distinct subtypes according to the distinct phenotype expression of CD45RA and CD25 among CD4+ T cells.[10] These include CD45RA+CD25+ FoxP3 low resting Treg cells (rTreg cells) and CD45RA-CD25+ FoxP3 high activated memory Treg cells (aTreg cells), which have been found to be suppressive in vitro, while cytokine-secreting CD45RA-CD25+ FoxP3 low population are non- suppressive T cells which retain the potential of differentiating into Th17 cells.[42] The blockade of the IL-21 pathway can significantly suppress IFN-γ and IL-17A secretion and increase FoxP3 expression in CD4+ T cells and thus restore the balance among Th1 and Th17 cells and Treg cells.[10] An elevated IL-6 levels can also promote the accumulation of CD4+ T cell independent of the accumulation of Treg cells, whereas the blockade of IL-6 receptor affects the imbalance causing an increase of Treg cells with activated phenotype but lead to a decrease of Treg cells with deficient functional phenotype.[44] On the contrary, glucocorticoids can effectively inhibit Th17 polarization rather than effectively rehabilitate the Treg cells deficiency observed in GCA.[16,44]
CD8+ T Cells in GCA
The CD8+ T cell is a stable participant in the pathogenesis of few important autoimmune diseases (including vacuities). The CD8+ T cells were previously considered to be associated with the disease progression in GCA.[45] McKinney, E. F. found that an expansion of circulating memory CD8+ T cells could correlate with the chance of relapse and poor prognosis in ANCA-associated vacuities (AAV) and systemic lupus erythematous.[46] Additionally, Subpopulations of CD8+ T cells could play a differential role in the development of GCA, including that of IFN-producing Tc1 cells, Tc2 cells, IL-17-producing Tc17 cells.[19]
Samson et al. elucidated that even when the percentage of Tc1 cells (CD3þCD8þIFN-γ) in GCA patients was similar to healthy controls, a higher percentage of circulating cytotoxic CD8+ T lymphocytes (CTL), Tc17 (CD3+CD8+IL-17+), CD63+CD8+ T cells and production of soluble granzymes A as well as B was observed in the patients (Figure 1). Their population also decreased significantly after the treatment with glucocorticoids.[47] The decreased circulating CD8+ T cells proportion has been associated with the clinical manifestation of GCA, such as carotid stenosis. GCA patients treated with GCs still maintain lower numbers of circulating CD8+ T cells, thus indicating the possibility of relapse or the requirement for long-term therapy with GCs.[47] Similarly, CD8+ T cells play a cytotoxic role in the lesion arteries by re-entering into the blood. CD8+ T cells with restricted TCR repertoire can be found in temporal artery biopsies (TABs), which can synthesize molecules such as granzyme B and anti-T-cell intra-cytoplasmic antigen (TiA1). The recruitment of CD8+ T cells can be triggered by CCL2, CXCL9, CXCL10 and CXCL11.[48-50] CD8+ T cells invade into arterial vessels, once activated by antigens and/or MHC classⅠchain-related gene A (MICA)[51] and transform into cytotoxic cytokine-producing cells.[48,49]
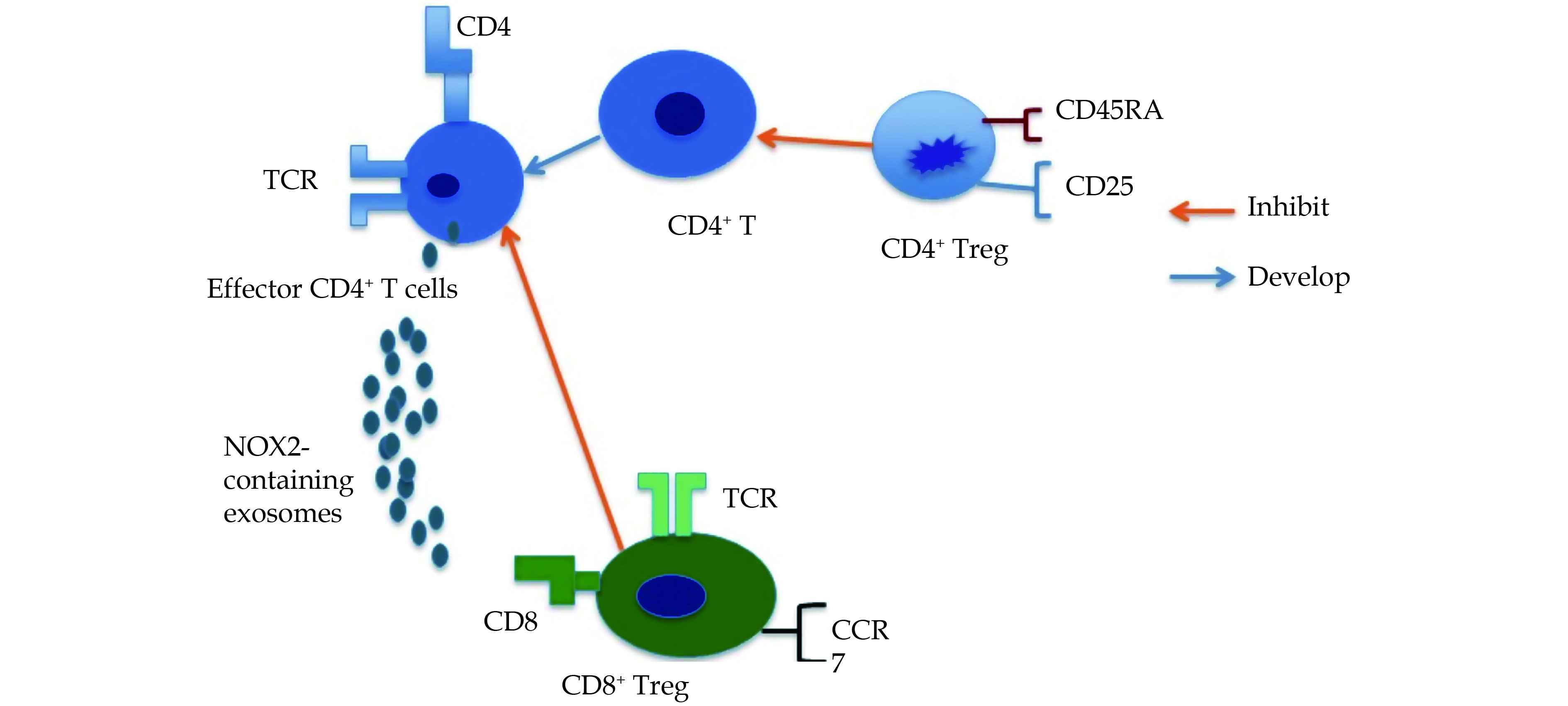
Figure
1.
The functions of regulatory T cells in GCA.
Both CD4+ and CD8+ Treg cells expressing FOXP3 can effectively inhibit the functions of effector CD4+ T cells. In addition, CD8+CCR7+ Tregs produce NOX-2-containing exosomes to suppress the potential role of effector CD4+ T cells. TCR: T-cell receptor.
In addition, the change in the numbers of circulating CTL is not consistent with change of erythrocyte sedimentation rate (ESR)and fibrinogen level. CD8+ T cells possess the ability to differentiate into Tc1, Tc2 and Tc17 cells besides their cytotoxic functions.[19,52] In GCA patients showing remission, Tc1 cells account for a large percentage of total CD8+ T cells and their number was not decreased by treatment with GCs, whereas the percentage of Tc17 cells was sensitive to GCs therapy. The levels of Tc17 cells can associate with acute phase reaction proteins including C-reactive protein (CRP), ESR and fibrinogen indicating.[10,11,16,47]
Apart from CD4+ Treg cells, CD8+ Treg cells are also involved in inhibiting and halting the immune responses which are tightly associated with chronic inflammation and autoimmune diseases.[53] A number of studies have revealed that specific the expression of CCR7 could effectively inhibit activation and expansion of CD4+ T cells dependent on the release of exosomes containing NADPH oxidase 2 (NOX2).[54] In young healthy individuals, CD8+ T cells can localize in the T cell zones of secondary lymphoid organs.
The CD8+ Treg cells in older individuals and GCA patients fail to exhibit significant immunosuppressive role in GCA because of deficiency of NADPH oxidase 2 (NOX2); a deficiency even more pronounced in GCA patients than in the healthy older individuals.[54] Membrane NOX2 is expressed selectively on CD8+ Treg cells rather than on the resting and activated CD8+ T cells.
Overexpression of NOX2 can also restores the suppressive functions of old CD8+ Treg cells, thereby suggesting that targeting oxidase to reinstate immune homeostasis can serve as a useful pharmacological strategy in the older individuals. At the molecular level, deficiency of NADPH oxidase 2 (NOX2) has been observed to drive CD8+ Treg cells impairment in the older individuals and in patients with GCA.[53,54]
T CELL SIGNALING IN GCA
TCR Signaling
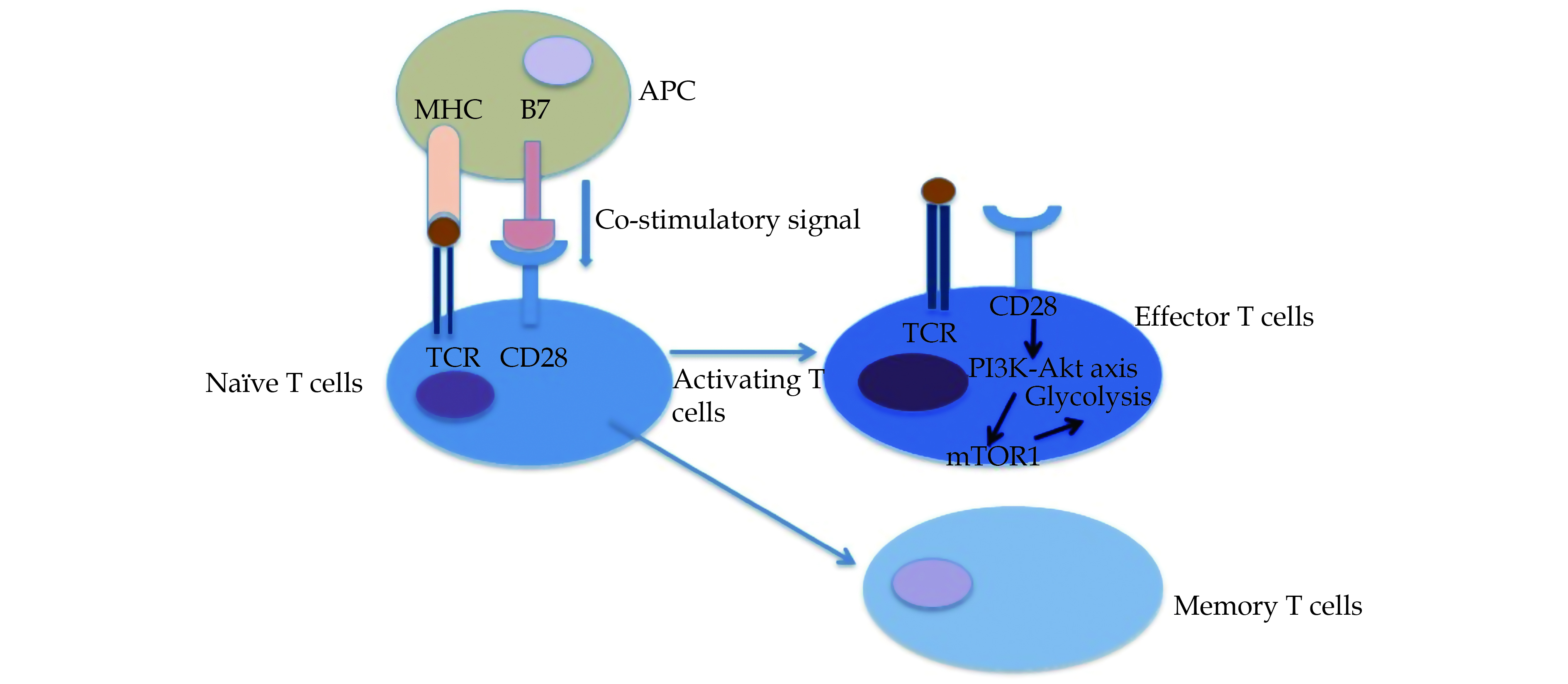
Aside from the recruitment of CD4+ T cells, T-cell activation also relies on the expression of co-stimulatory signals and the deficiency of co-inhibitory signals. Normally, CD4+ T cells do not exist in the arterial layers, but they can invade into the adventitia via vasa vacuum endothelial cells expressing vascular cell adhesion molecule (ICAM) such as ICAM-1 and VCAM-1 under pathological conditions.[55] Native CD4+ T cells can be activated by a combination of T cell receptor (TCR) with the peptide–major histocompatibility complex (MHC) and binding of stimulatory molecule (B7.1 or B7.2) to its receptor. The signals originating because of binding to TCR play a critical role in activating naive T cells (Figure 2). When TCR along with co-receptors CD28, binds to the peptide–MHC complex, it can effectively lead to the CD28 co-stimulation. The CD28 co-stimulation signals can stimulate the activation of the phosphatidylinositol 3-kinase (PI3K)-AKT axis and exemplify flux of glycolysis.[56,57] Activated effector T cells require energy meet the demand of cell clonal expansion and differentiation. ATP synthesis is critical to maintain the support of energy, mainly by virtue of glycolytic pathway.[58] In the metabolic process of glycolysis, mechanistic target of rapamycin complex (mTOR), in particular mTOR1 (part of mTOR complex 1) regulates the differentiation and functions of effector T cells via integrating micro-environmental considerations. In GCA, mTORC1 has been reported to be continuously active. In a GCA mouse model embedded with human arteries, blocking CD28 effectively inhibited mTORC1 activation, reduced the efficiency of T cell energy utilization and subsequently down-regulated aberrant inflammatory response.[43]
Figure
2.
TCR combines with peptide–MHC and CD28 binds to B7 on the surface of APC.
The CD28-modulating signaling activates mTOR1 pathway via PI3K-Akt axis, thus regulating glycolysis and causing differentiation of CD4+ T cells into effector T cells and memory T cells. APC: antigen presenting cells; TCR: T-cell receptor.
CD4 T cells differentiation towards their subtypes is primarily determined by the cytokine mileu. TGF-β signals play an important role in polarization of CD4 T cells. At an early stage, TGF-β can regulate generation of both Th17 and Treg cells.[59] However, the further polarization requires the presence of IL-6 or IL-2. T cells cytokines receptors communicate with cytokine milieu, depending on the Janus kinase-signal transducer of activators of transcription (JAK-STAT) signals, thereby triggering differentiation of CD4+ T cells into Th17 cells and Th1 cells.[60,61] Zhang et al detected a significant increase in expression of few STAT-dependent target genes, including T-bet, CXCL9, ISG15, OAS1, distinguishable from normal individuals and patients with vacuities.[20] Both type I and II interferon(IFN I and IFN II) can induce activation of JAK-STAT pathway. In GCA, the level of STAT1 and STAT2 was reported to be high, while the expression of STAT3 and STAT5 was observed to be at a lower level. STAT3 also plays a suppressive role in Treg cell differentiation by modulating the expression of Foxp3.[43] Additionally, the phosphorylation and activation STAT3 can drive Th17-specific genes, such as Rorc, Il17, and Il23r, thus promoting the differentiation of CD4 T cells into Th17 cells.[62] TGF-β and IL-2 can induce Treg cell differentiation by phosphorylating STAT5 and promoting the expression of Foxp3.[63] However, TGF-β was found to inhibit the polarization of CD4 T cells towards Th1 and Th2 cells, and IL-2 can suppress the differentiation of Th17 cells.[64,65]
Effector CD4 T cells lack the mTOR complex (mTORC, including mTORC1 and mTORC2), leading to dysfunction of effector T cells and transformation into Tregs.[66] Insufficient mTOR activity and inhibition of the mTORC activity by rapamycin has been found to promote expansion of Treg cells through affecting different pathways. Both JAK-STAT and CD28-PI3K-AKT pathways obviously influence the balance of Th17/Treg ratio. Insufficient mTORC activity can improve the susceptibility of T cells to TGF-β and rapamycin can stimulate Foxp3 expression. mTORC1 induced HIF1-α protein acts to stimulate the glycolytic pathway and promote the expression of RAR-related orphan receptor (ROR)γt to promote the differentiation of Th17 cells, whereas it can also suppress the expression of Foxp3 to down regulate Th17 cells differentiation.
The importance of JAK- STAT signaling in GCA can be supported by significant inhibitory results obtained with Tofacitinib, a substantial down regulation of lineage dependent transcription factors (T-bet, RORC and BCL-6) and T-cell effector molecules (IFN- γ, IL-17 and IL-21).[20]
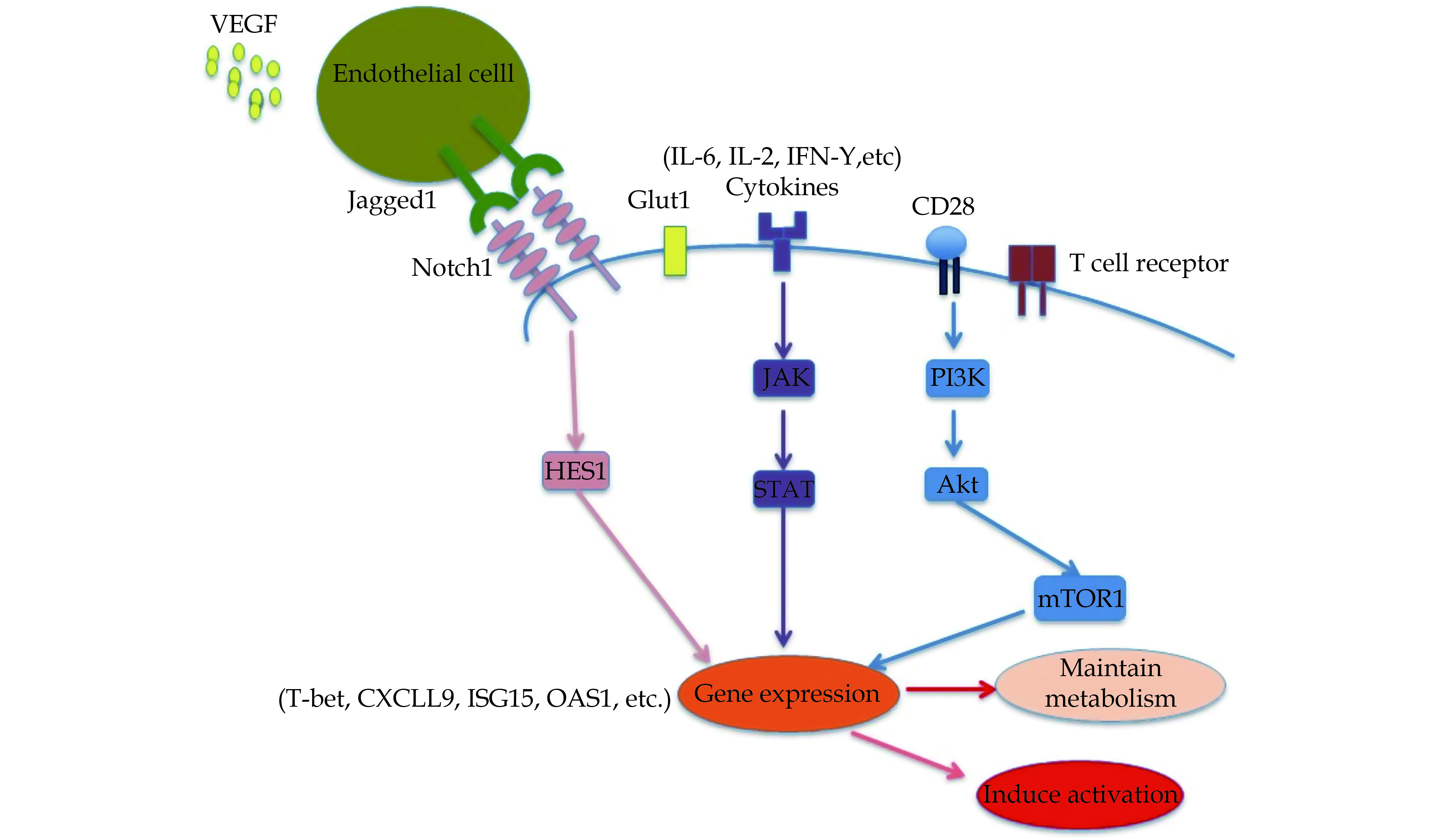
VEGF-Jagged-Notch Signaling
In GCA patients, the circulating level of vascular endothelial growth factor (VEGF) is overexpressed by Endothelial cells (ECs,) thereby triggering the production of Notch ligand Jagged1 (Figure 3). Notch is involved in the differentiation through affecting the production of T-bet as well as RORgt, and thus functioning as lineage-defining transcription factors. T effector cell activity could also be modulated by Jagged-Notch signaling.[22] Overall, the activation of mTORC1 can be induced by CD28-PI3K-AKT axis as well as Jagged1-Notch1 pathway; both of CD28-PI3K-AKT axis and Jagged1-Notch1 pathway can contribute to the polarization of Th1 and Th17 with special pathogenic effects.[67]
Figure
3.
Signaling pathways of TCR, stimulatory signal, cytokine signal and JAK-STAT signal.
CD28-dependent co-stimulatory signals induce activation of mTOR1; resulting in an effector T cells lineage formation and thus regulates the metabolism of T cells. VEGF-Jagged-Notch axis is the symbol of abnormality of pathogenic T cells. JAK-STAT signaling is involved in the regulation of the various functions of T cells by stimulating the production of different cytokines. TCR: T-cell receptor; VEGF: vascular endothelial growth factor.
In addition, the interaction between CD4+ T cells expressing Notch1 and ECs expressing Jagged-1, a ligand for the Notch1 receptor, can enable CD4+T cells to enter into the adventitia and actively regulate the differentiation of CD4+ T cells into different subpopulation. Further, increased VEGF levels in the circulation of patients with GCA can promote the aberrant expression of Jagged-1 on adventitial micro vascular ECs through modulating mTOR pathway, which enables the EC to effectively play a stimulatory role in inducing CD4+ T cells differentiation. In GCA patients, an increased expression of surface Notch1 receptor and Notch target protein HES1 has been found. In addition, the expression of Notch1 associated with HES1 has been reported to be tightly regulated in CD4 T cells of individual patients.[22]
Except for CD4T+cells, the communication between CD4T+cells and dendritic cells or macrophages (as instrumental cellular participators) has also been found to be involved in the pathogenesis of GCA.
T CELLS-DENDRITIC CELLS INTERPLAY
Dendritic cells primarily act as the trigger of arteritis by producing inflammatory cytokines (in particular, IL-6 and IL-18) and express CD86 to initiate and regulate the arteritis. The activating signal of CD4+ T cells is provided by dendritic cells (DCs) expressing Toll-like receptors (TLRs) in the adventitia, where antigen can actively invade into an artery. In healthy arteries, adventitial DCs have been found to be inactivated and are thus unable to communicate with all reactive T cells. Upon the stimulation of TLRs via pathogen-associated molecular patterns (PAMPs) or danger associated molecular patterns (DAMPs), DCs subsequently recruit, accumulate, activate and mature inside the arteries. Mature DCs can then migrate into media; secrete chemokine and express CD83 and CD86 antigens, thereby resulting in the recruitment of T cells. Anti-CD83 antibodies can significantly decrease the level of IFN-γ and IL-1β in temporal artery of SCID mouse chimeras. DCs recruiting T cells could be induced by the expression of chemokine, in particular CCL20. Moreover, DCs expressing CCL20 can also drive the recruitments of CD4+ T cells via binding to CCL20 ligand, CCR6.
In addition, once CD4+ T cells enter into the wall, CD4+ T cells can communicate with dendritic cells (vasDC) located in the vessels, and lack the expression of immune checkpoint proteins including Programmed death-1 (PD-1).[68] PD-1 provided inhibitory signals to T cells through the binding to cell death ligand 1 and 2 (PD-L1 and PD-L2), thus leading to T cells apoptosis and even differentiation towards Treg cells.[69] Once the antigens have been recognized by the DCs, they can only send the co-stimulatory signal to T cells through the direct interactions between T-cell receptor and DCs. In GCA, the impaired function of co-inhibitory signals can trigger expansion of the effector T cells and the production of multiple cytokine.[70] Considering that inhibitory checkpoints are ineffective in GCA therapy, CD28-dependent stimulation pathway seems to play a primary catalytic role in the pathogenic process of GCA.[71]
Therefore, DCs only provide co-stimulatory signals and lack sufficient co-inhibitory signals for T cells but vasDCs can recognize T cell signals and promote their in situ activation in GCA. Therefore, DCs are primarily involved in the recruitment and activation of T cells in GCA.
T CELLS-MACROPHAGE INTERPLAY
Macrophages along with CD4+ T cells are the main constituents of the granulomatous lesions. Once monocytes differentiate into the macrophages, they trigger the synthesis of chemokine (CXCL9, 10, 11, CCL5, and CCL 18) and pro-inflammatory cytokines (IL-6, IL-1β and TNF-α) and drive the local inflammatory cascade.[72] In GCA, macrophages can regulate the secretion of T cell chemoattractants (CXCL9, CXCL10) but not that of IL-1β and IL-6, and these cytokine-secreting macrophages were specifically low for PD-L1 expression.[73] Additionally, stimulated medial macrophages are also responsible for the synthesis of nitric oxide (NO) and matrix metalloproteinase (MMPs), including MMP-2, MMP-7, and MMP-9. NO actively contributes to the matrix degradation and MMPs, particularly MMP-9 secreted by CD68+ macrophages can induce the destruction of artery media and internal elastic lamina, thereby facilitating the movement of T cells and vascular smooth muscle cells (VSMCs) into intima.[37,72]
In GCA, macrophages can act as pro-inflammatory effector cells, while in coronary artery disease (CAD) they can also generate anti-inflammatory signals due to the up-regulated expression of PD-L1 mediated by extracellular glucose concentrations.[73] The pro-inflammatory functions of macrophages primarily depend on the high efficiency of glucose utilization, which appears to be the case in the macrophages from the healthy individuals.[67] However, lower expression of PD-L1 undermines the anti-inflammatory role of macrophages in GCA; and when macrophages invade into microenvironments, they can gain other non-glucose energy sources thereby resulting in prolonged persistence in local lesions and continuous inflammation of artery wall.
IL12/TH1/IFN-Γ PATHWAY
Indeed, IL-12/Th1/IFN-γ pathway has been reported to actively associate with the local vessel inflammation in GCA, especially the neuro-ophthalmic ischemic complications.[7,74] A number of studies have demonstrated that IL-12 was absent in the healthy individuals, while its level was elevated in the serum of untreated GCA patients, particularly these patients displaying cranial ischemic complications irrespective of their treatment with GCs.[15] Consistent with these findings, an elevated gene expression of IL-12p35 has been reported in the monocytes of GCA patients, rather than healthy controls.[15]
Moreover, the transcriptome study of arteries in patients with GCA has unveiled the abundance of type I and II IFN in the arterial lesions.[20] The type I IFN-producing cells has not been elucidated yet. Type I IFN may also provide a bridge between the innate and the adaptive immune responses.[75]
IFN-γ participates in the migration and proliferation of VSMCs, and function to promote micro vascular angiogenesis. One ex vivo trial has demonstrated that IFN-γ exerted effects on VSMCs through amplification of synthesis of various chemokines such as CCL5, CXCL9, CXCL10, CXCL11, CX3CL1 and adhesion molecules by VSMCs, thereby recruiting monocytes and resulting in the formation of multinucleated giant cells.[48]
IFN-γ may also be involved in the loss of lesion vessel medial layer via triggering apoptosis of VSMCs dependent on STAT3 signaling.[7] Similar results were obtained in the gene knockdown (including that of the pro-apoptotic molecules X-linked inhibitor of apoptosis associated factor-1 (XAF1) and Noxa chimeric animals of immune-deficient mice embedded with human coronary artery grafts.[76] IFN-γ-induced STAT3 signaling in VSMCs effectively triggered inhibition of STAT1 activation and promoted the apoptosis of VSMCs by virtue of both death receptor and mitochondrial-mediated pathways.[76] Therefore, the pleiotropic effects of IFN-γ on VSMCs mediated through JAK-STAT signaling may deserve advanced exploration.
IL-32 is another effector cytokine triggered by IFN-γ, which can correlate with the index of clinical disease activity.[77] The stimulation to CD4+ T cells with IL-21 promoted Th1 and Th17 cell expansion, and blockade of IL-21 using IL-21R-Fc significantly decreased the secretion of IL-17A and IFN-γ.[10] IFN-γ has been found to be less responsive to the treatment of corticosteroids than other cytokines.[11] Once the dosing of GCs has been tapered or when GCA patients discontinued the use of GCs, the relapse of disease was more prone to occur possibly due to an incomplete inhibition of GCs on Th1 cells.[48]
CONCLUSIONS
The pathology of GCA is complicated and not completely understood. Adaptive immune response especially the T cell adaptive immunity exerts a crucial role in the pathogenesis of GCA, including the development of vascular inflammation; insufficient immunosuppression in T cell immune response can correlate with the formation of granulomas in the arterial wall. In this article, we have reviewed the involvement of different signaling pathways, as well as that of several immune T cells subsets and their effector cytokines in the immunopathology of GCA, which can form the basis of novel therapy against GCA. The role of T cells in GCA requires collaboration of multiple cellular signaling cascades, among which the continuous activation of TCR signaling, VEGF-Jagged-Notch signaling and the JAK-STAT pathways have been found to play an extremely important role. With the more advanced knowledge of immunology in GCA, a multitude of novel factors can be detected and more effective clinical interventions can be applied to the control the disease progression and promote an effective clinical remission.
ACKNOWLEDGEMENTS
This study was supported by National Natural Science Foundation of China (Number: 8187061400).
Blockmans DE. Polymyalgia rheumatica and giant-cell arteritis. N Engl J Med 2002; 347: 2083−2085. doi: 10.1056/NEJM200212193472521
[2]
Espitia O, Samson M, Le Gallou T, et al. Comparison of idiopathic (isolated) aortitis and giant cell arteritis-related aortitis. A French retrospective multicenter study of 117 patients. Autoimmun Rev 2016; 15: 571−576.
[3]
Berti A, Campochiaro C, Cavalli G, et al. Giant cell arteritis restricted to the limb arteries: An overlooked clinical entity. Autoimmun Rev 2015; 14: 352−357. doi: 10.1016/j.autrev.2014.12.005
[4]
Gonzalez-Gay MA, Miranda-Filloy JA, Lopez-Diaz MJ, et al. Giant cell arteritis in northwestern Spain: a 25-year epidemiologic study. Medicine 2007; 86: 61−68. doi: 10.1097/md.0b013e31803d1764
[5]
Wu CS, Hsu KL, Chang YL, et al. Giant cell arteritis with CD8+ instead of CD4+ T lymphocytes as the predominant infiltrating cells in a young woman. J Microbiol Immunol Infect 2004; 37: 246−249.
[6]
Carmona FD, Vaglio A, Mackie SL, et al. A genome-wide association study identifies risk alleles in plasminogen and P4HA2 associated with giant cell arteritis. Am J Hum Genet 2017; 100: 64−74. doi: 10.1016/j.ajhg.2016.11.013
[7]
Weyand CM, Goronzy JJ. Immune mechanisms in medium and large-vessel vasculitis. Nat Rev Rheumatol 2013; 9: 731−740. doi: 10.1038/nrrheum.2013.161
[8]
Deng J, Ma-Krupa W, Gewirtz AT et al. Toll-like receptors 4 and 5 induce distinct types of vasculitis. Circ Res 2009; 104: 488−495. doi: 10.1161/CIRCRESAHA.108.185777
Terrier B, Geri G, Chaara W, et al. Interleukin-21 modulates Th1 and Th17 responses in giant cell arteritis. Arthritis & Rheumatism 2012; 64: 2001−2011.
[11]
Deng J, Younge BR, Olshen RA, et al. Th17 and Th1 T-cell responses in giant cell arteritis. Circulation 2010; 121: 906−915. doi: 10.1161/CIRCULATIONAHA.109.872903
[12]
Salvarani C, Pipitone N, Versari A, et al. Clinical features of polymyalgia rheumatica and giant cell arteritis. Nat Rev Rheumatol 2012; 8: 509−521. doi: 10.1038/nrrheum.2012.97
[13]
Weyand CM, Watanabe R, Zhang H, et al. Cytokines, growth factors and proteases in medium and large vessel vasculitis. Clin Immunol (Orlando, Fla) 2019; 206: 33−41. doi: 10.1016/j.clim.2019.02.007
[14]
Nakanishi K, Yoshimoto T, Tsutsui H, et al. Interleukin-18 regulates both Th1 and Th2 responses. Annu Rev Immunol 2001; 19: 423−474. doi: 10.1146/annurev.immunol.19.1.423
[15]
Conway R, O'Neill L, McCarthy GM, et al. Interleukin 12 and interleukin 23 play key pathogenic roles in inflammatory and proliferative pathways in giant cell arteritis. Ann Rheum Dis 2018; 77: 1815−1824. doi: 10.1136/annrheumdis-2018-213488
[16]
Samson M, Audia S, Fraszczak J, et al. Th1 and Th17 lymphocytes expressing CD161 are implicated in giant cell arteritis and polymyalgia rheumatica pathogenesis. Arthritis Rheum 2012; 64: 3788−3798. doi: 10.1002/art.34647
[17]
Lee YK, Mukasa R, Hatton RD, et al. Developmental plasticity of Th17 and Treg cells. Curr Opin Immunol 2009; 21: 274−280. doi: 10.1016/j.coi.2009.05.021
[18]
Murphy KM, Ouyang W, Farrar JD, et al. Signaling and transcription in T helper development. Annu Rev Immunol 2000; 18: 451−494. doi: 10.1146/annurev.immunol.18.1.451
[19]
Annunziato F, Romagnani C, Romagnani S. The 3 major types of innate and adaptive cell-mediated effector immunity. J Allergy Clin Immunol 2015; 135: 626−635. doi: 10.1016/j.jaci.2014.11.001
[20]
Zhang H, Watanabe R, Berry GJ, et al. Inhibition of JAK-STAT signaling suppresses pathogenic immune responses in medium and large vessel vasculitis. Circulation 2018; 137: 1934−1948. doi: 10.1161/CIRCULATIONAHA.117.030423
[21]
Yu L, Qin L, Zhang H, et al. AIP1 prevents graft arteriosclerosis by inhibiting interferon-gamma-dependent smooth muscle cell proliferation and intimal expansion. Circ Res 2011; 109: 418−427. doi: 10.1161/CIRCRESAHA.111.248245
[22]
Wen Z, Shen Y, Berry G, et al. The microvascular niche instructs T cells in large vessel vasculitis via the VEGF-Jagged1-Notch pathway. Sci Transl Med 2017; 9: eaal3322. doi: 10.1126/scitranslmed.aal3322
[23]
Bonecchi R, Bianchi G, Bordignon PP, et al. Differential expression of chemokine receptors and chemotactic responsiveness of type 1 T helper cells (Th1s) and Th2s. J Exp Med 1998; 187: 129−134. doi: 10.1084/jem.187.1.129
[24]
Ciccia F, Alessandro R, Rizzo A, et al. IL-33 is overexpressed in the inflamed arteries of patients with giant cell arteritis. Ann Rheum Dis 2013; 72: 258−264. doi: 10.1136/annrheumdis-2012-201309
[25]
Blom L, Poulsen BC, Jensen BM, et al. IL-33 induces IL-9 production in human CD4+ T cells and basophils. PloS one 2011; 6: e21695. doi: 10.1371/journal.pone.0021695
Ciccia F, Rizzo A, Guggino G, et al. Difference in the expression of IL-9 and IL-17 correlates with different histological pattern of vascular wall injury in giant cell arteritis. Rheumatology (Oxford, England) 2015; 54: 1596−1604. doi: 10.1093/rheumatology/kev102
[28]
Abdelilah S, Latifa K, Esra N, et al. Functional expression of IL-9 receptor by human neutrophils from asthmatic donors: role in IL-8 release. J Immunol 2001; 166: 2768−2774. doi: 10.4049/jimmunol.166.4.2768
[29]
Ciccia F, Rizzo A, Ferrante A, et al. New insights into the pathogenesis of giant cell arteritis. Autoimmun Rev 2017; 16: 675−683. doi: 10.1016/j.autrev.2017.05.004
[30]
Weyand CM, Younge BR, Goronzy JJ. IFN-gamma and IL-17: the two faces of T-cell pathology in giant cell arteritis. Curr Opin Rheumatol 2011; 23: 43−49. doi: 10.1097/BOR.0b013e32833ee946
Langley RG, Elewski BE, Lebwohl M, et al. Secukinumab in plaque psoriasis--results of two phase 3 trials. N Engl J Med 2014; 371: 326−338. doi: 10.1056/NEJMoa1314258
[33]
Watanabe R, Hosgur E, Zhang H, et al. Pro-inflammatory and anti-inflammatory T cells in giant cell arteritis. Joint Bone Spine 2017; 84: 421−426. doi: 10.1016/j.jbspin.2016.07.005
[34]
Espígol-Frigolé G, Corbera-Bellalta M, Planas-Rigol E, et al. Increased IL-17A expression in temporal artery lesions is a predictor of sustained response to glucocorticoid treatment in patients with giant-cell arteritis. Ann Rheum Dis 2013; 72: 1481−1487. doi: 10.1136/annrheumdis-2012-201836
[35]
Samson M, Audia S, Janikashvili N, et al. Are IL-10+ regulatory Th17 cells implicated in the sustained response to glucocorticoid treatment in patients with giant cell arteritis? Comment on the paper of Espigol-Frigole et al. Ann Rheum Dis 2013; 72: e3. doi: 10.1136/annrheumdis-2013-203439
[36]
Annunziato F, Cosmi L, Liotta F, et al. Defining the human T helper 17 cell phenotype. Trends Immunol 2012; 33: 505−512. doi: 10.1016/j.it.2012.05.004
[37]
Watanabe R, Maeda T, Zhang H, et al. MMP (Matrix Metalloprotease)-9-producing monocytes enable T cells to invade the vessel wall and cause vasculitis. Circ Res 2018; 123: 700−715. doi: 10.1161/CIRCRESAHA.118.313206
[38]
Korn T, Bettelli E, Gao W, et al. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature 2007; 448: 484−487. doi: 10.1038/nature05970
[39]
Vogelzang A, McGuire HM, Yu D, et al. A fundamental role for interleukin-21 in the generation of T follicular helper cells. Immunity 2008; 29: 127−137. doi: 10.1016/j.immuni.2008.06.001
[40]
Ettinger R, Sims GP, Fairhurst AM, et al. IL-21 induces differentiation of human naive and memory B cells into antibody-secreting plasma cells. J Immunol 2005; 175: 7867−7879. doi: 10.4049/jimmunol.175.12.7867
[41]
Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol 2012; 30: 531−564. doi: 10.1146/annurev.immunol.25.022106.141623
[42]
Miyara M, Yoshioka Y, Kitoh A, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity 2009; 30: 899−911. doi: 10.1016/j.immuni.2009.03.019
[43]
Lee GR. The Balance of Th17 versus Treg Cells in Autoimmunity. Int J Mol Sci 2018; 19: 730. doi: 10.3390/ijms19030730
[44]
Miyabe C, Miyabe Y, Strle K, et al. An expanded population of pathogenic regulatory T cells in giant cell arteritis is abrogated by IL-6 blockade therapy. Ann Rheum Dis 2017; 76: 898−905. doi: 10.1136/annrheumdis-2016-210070
[45]
Martinez-Taboada VM, Blanco R, Fito C, et al. Circulating CD8+ T cells in polymyalgia rheumatica and giant cell arteritis: a review. Semin Arthritis Rheum 2001; 30: 257−271. doi: 10.1053/sarh.2001.9734
[46]
McKinney EF, Lyons PA, Carr EJ, et al. A CD8+ T cell transcription signature predicts prognosis in autoimmune disease. Nat Med 2010; 16: 586−591. doi: 10.1038/nm.2130
[47]
Samson M, Ly KH, Tournier B, et al. Involvement and prognosis value of CD8+ T cells in giant cell arteritis. J Autoimmun 2016; 72: 73−83. doi: 10.1016/j.jaut.2016.05.008
[48]
Corbera-Bellalta M, Planas-Rigol E, et al. Blocking interferon gamma reduces expression of chemokines CXCL9, CXCL10 and CXCL11 and decreases macrophage infiltration in ex vivo cultured arteries from patients with giant cell arteritis. Ann Rheum Dis 2016; 75: 1177−1186. doi: 10.1136/annrheumdis-2015-208371
[49]
Antonelli A, Ferrari SM, Giuggioli D, et al. Chemokine (C-X-C motif) ligand (CXCL)10 in autoimmune diseases. Autoimmun Rev 2014; 13: 272−280. doi: 10.1016/j.autrev.2013.10.010
[50]
Muller M, Carter S, Hofer MJ, et al. Review: The chemokine receptor CXCR3 and its ligands CXCL9, CXCL10 and CXCL11 in neuroimmunity--a tale of conflict and conundrum. Neuropathol Appl Neurobiol 2010; 36: 368−387. doi: 10.1111/j.1365-2990.2010.01089.x
[51]
Dejaco C, Duftner C, Al-Massad J, et al. NKG2D stimulated T-cell autoreactivity in giant cell arteritis and polymyalgia rheumatica. Ann Rheum Dis 2013; 72: 1852−1859. doi: 10.1136/annrheumdis-2012-201660
[52]
Arens R, Schoenberger SP. Plasticity in programming of effector and memory CD8 T-cell formation. Immunol Rev 2010; 235: 190−205. doi: 10.1111/j.0105-2896.2010.00899.x
[53]
Collison J. Vasculitis syndromes: Dysfunctional CD8 TREG cells implicated in GCA. Nat Rev Rheumatol 2016; 12: 314.
[54]
Wen Z, Shimojima Y, Shirai T, et al. NADPH oxidase deficiency underlies dysfunction of aged CD8+ Tregs. J Clin Invest 2016; 126: 1953−1967. doi: 10.1172/JCI84181
[55]
Samson M, Corbera-Bellalta M, Audia S, et al. Recent advances in our understanding of giant cell arteritis pathogenesis. Autoimmun Rev 2017; 16: 833−844. doi: 10.1016/j.autrev.2017.05.014
[56]
Kane LP, Weiss A. The PI-3 kinase/Akt pathway and T cell activation: pleiotropic pathways downstream of PIP3. Immunol Rev 2003; 192: 7−20. doi: 10.1034/j.1600-065X.2003.00008.x
[57]
Frauwirth KA, Riley JL, Harris MH, et al. The CD28 signaling pathway regulates glucose metabolism. Immunity 2002; 16: 769−777. doi: 10.1016/S1074-7613(02)00323-0
[58]
MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Annu Rev Immunol 2013; 31: 259−283. doi: 10.1146/annurev-immunol-032712-095956
[59]
Manel N, Unutmaz D, Littman DR. The differentiation of human T(H)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgammat. Nat immunol 2008; 9: 641−649. doi: 10.1038/ni.1610
[60]
O’Shea JJ, Plenge R. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity 2012; 36: 542−550. doi: 10.1016/j.immuni.2012.03.014
[61]
Mullen AC, High FA, Hutchins AS, et al. Role of T-bet in commitment of TH1 cells before IL-12-dependent selection. Science 2001; 292: 1907−1910. doi: 10.1126/science.1059835
[62]
Durant L, Watford WT, Ramos HL, et al. Diverse targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis. Immunity 2010; 32: 605−615. doi: 10.1016/j.immuni.2010.05.003
[63]
Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science 2003; 299: 1057−1061. doi: 10.1126/science.1079490
[64]
Leentjens J, Bekkering S, Joosten LAB, et al. Trained innate immunity as a novel mechanism linking infection and the development of atherosclerosis. Circ Res 2018; 122: 664−669. doi: 10.1161/CIRCRESAHA.117.312465
[65]
Quintin J, Cheng SC, van der Meer JW, et al. Innate immune memory: towards a better understanding of host defense mechanisms. Curr Opin Immunol 2014; 29: 1−7. doi: 10.1016/j.coi.2014.02.006
[66]
Barbi J, Pardoll D, Pan F. Metabolic control of the Treg/Th17 axis. Immunol Rev 2013; 252: 52−77. doi: 10.1111/imr.12029
[67]
Watanabe R, Berry GJ, Liang DH, et al. Cellular signaling pathways in medium and large vessel vasculitis. Front Immunol 2020; 11: 587089. doi: 10.3389/fimmu.2020.587089
[68]
Weyand CM, Berry GJ, Goronzy JJ. The immunoinhibitory PD-1/PD-L1 pathway in inflammatory blood vessel disease. J Leukoc Biol 2018; 103: 565−575.
[69]
Rosenblatt J, Glotzbecker B, Mills H, et al. PD-1 blockade by CT-011, anti-PD-1 antibody, enhances ex vivo T-cell responses to autologous dendritic cell/myeloma fusion vaccine. J Immunother 2011; 34: 409−418. doi: 10.1097/CJI.0b013e31821ca6ce
[70]
Zhang H, Watanabe R, Berry GJ, et al. Immunoinhibitory checkpoint deficiency in medium and large vessel vasculitis. Proc Natl Acad Sci U S A 2017; 114: E970−E979. doi: 10.1073/pnas.1616848114
[71]
Langford CA, Cuthbertson D, Ytterberg SR, et al. A randomized, double-blind trial of abatacept (CTLA-4Ig) for the treatment of giant cell arteritis. Arthritis Rheumatol 2017; 69: 837−845. doi: 10.1002/art.40044
[72]
Guevara M, Kollipara CS. Recent advances in giant cell arteritis. Curr Rheumatol Rep 2018; 20: 25. doi: 10.1007/s11926-018-0737-1
[73]
Watanabe R, Hilhorst M, Zhang H, et al. Glucose metabolism controls disease-specific signatures of macrophage effector functions. JCI Insight 2018: 3.
[74]
Kermani TA. Takayasu arteritis and giant cell arteritis: are they a spectrum of the same disease? Int J Rheum Dis 2019; 22(Suppl 1): S41−S48.
[75]
Nordborg C, Larsson K, Aman P, et al. Expression of the class I interferon-related MxA protein in temporal arteries in polymyalgia rheumatica and temporal arteritis. Scand J Rheumatol 2009; 38: 144−148. doi: 10.1080/03009740802448841
[76]
Bai Y, Ahmad U, Wang Y, et al. Interferon-gamma induces X-linked inhibitor of apoptosis-associated factor-1 and Noxa expression and potentiates human vascular smooth muscle cell apoptosis by STAT3 activation. J Biol Chem 2008; 283: 6832−6842. doi: 10.1074/jbc.M706021200
[77]
Ciccia F, Alessandro R, Rizzo A, et al. Expression of interleukin-32 in the inflamed arteries of patients with giant cell arteritis. Arthritis Rheum 2011; 63: 2097−2104. doi: 10.1002/art.30374


 DownLoad:
DownLoad: