Please cite this article as: ZHANG Y, LIU MH, ZHANG M, WU GX, LIU J, WANG JZ, SUN XL, JIANG W, WANG D, KANG LM, WU XY, ZOU YB, SONG L. Different clinical characteristics and outcomes of hypertrophic cardiomyopathy with and without hypertension: seeking the truth. J Geriatr Cardiol 2023; 20(2): 109−120. DOI: 10.26599/1671-5411.2023.02.007.
Citation:
Please cite this article as: ZHANG Y, LIU MH, ZHANG M, WU GX, LIU J, WANG JZ, SUN XL, JIANG W, WANG D, KANG LM, WU XY, ZOU YB, SONG L. Different clinical characteristics and outcomes of hypertrophic cardiomyopathy with and without hypertension: seeking the truth. J Geriatr Cardiol 2023; 20(2): 109−120. DOI: 10.26599/1671-5411.2023.02.007.
Please cite this article as: ZHANG Y, LIU MH, ZHANG M, WU GX, LIU J, WANG JZ, SUN XL, JIANG W, WANG D, KANG LM, WU XY, ZOU YB, SONG L. Different clinical characteristics and outcomes of hypertrophic cardiomyopathy with and without hypertension: seeking the truth. J Geriatr Cardiol 2023; 20(2): 109−120. DOI: 10.26599/1671-5411.2023.02.007.
Citation:
Please cite this article as: ZHANG Y, LIU MH, ZHANG M, WU GX, LIU J, WANG JZ, SUN XL, JIANG W, WANG D, KANG LM, WU XY, ZOU YB, SONG L. Different clinical characteristics and outcomes of hypertrophic cardiomyopathy with and without hypertension: seeking the truth. J Geriatr Cardiol 2023; 20(2): 109−120. DOI: 10.26599/1671-5411.2023.02.007.
State Key Laboratory of Cardiovascular Disease, Fuwai Hospital, National Center for Cardiovascular Diseases, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China
2.
Cardiomyopathy Ward, Fuwai Hospital, National Center for Cardiovascular Disease, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China
3.
Department of Cardiology, Fuwai Hospital, National Center for Cardiovascular Disease, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China
4.
National Clinical Research Center of Cardiovascular Diseases, Fuwai Hospital, National Center for Cardiovascular Diseases, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China
OBJECTIVE To determine the different clinical characteristics and outcomes of hypertrophic cardiomyopathy (HCM) patients with and without hypertension (HT).
METHODS A total of 696 HCM patients were included in this study and all HCM diagnoses were confirmed by the genetic test. Patients were analyzed separately in the septal reduction therapy (SRT) cohort and the non-SRT cohort. The primary endpoint was cardiovascular death and the secondary endpoint was all-cause death. Outcome analyses were conducted to evaluate the associations between HT and outcomes in HCM. Medications before enrollment and at discharge were collected in the post-hoc analyses.
RESULTS HCM patients without HT were younger, had a lower body mass index, were more likely to have a family history of HCM, and had a smaller left ventricular (LV) end-diastolic diameter than those with HT in both cohorts. A thicker LV wall, a higher level of N-terminal pro-B-type natriuretic peptide, and a higher extent of LV late gadolinium enhancement were additionally observed in patients without HT in the non-SRT cohort. The presence of HT did not alter the distribution pattern of late gadolinium enhancement, as well as the constituent ratio of eight disease-causing sarcomeric gene variants in both cohorts. Outcome analyses showed that in the non-SRT cohort, patients without HT had higher risks of cardiovascular death (HR = 2.537, P = 0.032) and all-cause death (HR = 3.309, P = 0.032). While such prognostic divergence was not observed in the SRT cohort. Further post-hoc analyses in the non-SRT cohort found that patients without HT received fewer non-dihydropyridine calcium channel blockers and angiotensin-converting enzyme inhibitors/angiotensin receptor blockers before enrollment and at discharge.
CONCLUSIONS HCM patients without HT had worse clinical conditions and higher mortality than patients with HT overall, which may result from active medical therapy in HT patients. Active SRT may have a substantial de-risking effect on patients meeting the indications.
Hypertrophic cardiomyopathy (HCM) is predominantly characterized by the presence of left ventricular (LV) hypertrophy, decreased compliance, and myocardial fibrosis.[1] HCM is typically diagnosed by the extent of myocardial hypertrophy on imaging techniques.[2] Hypertension (HT) is one of the most common cardiovascular diseases and results in heavy burdens worldwide.[3] Although the co-occurrence of HCM and HT is common in clinical practice, no clear conclusions have been drawn on how HT should be regarded in the setting of HCM to date. Given that isolated HT can also lead to such hypertrophic myocardium mimicking HCM,[4] the investigations of the comorbid effects of HT on HCM should be based on the premise that the studied patients were all primary HCM. However, previous differential diagnostic strategies between primary HCM and isolated hypertensive cardiac hypertrophy are often empirical, and this unclear diagnosis of HCM in hypertensive patients prevented the ability to determine the true implications of this comorbid condition in the previous studies.[5,6]
Septal reduction therapy (SRT), including myectomy and alcohol septal ablation, are the preferred approaches to relieve severe hemodynamic compromise and improve symptoms in HCM patients, while is rarely indicated for asymptomatic patients with normal hemodynamics.[7–9] Patients who undergo SRT always have a large pathophysiological and prognostic gap compared with those who do not, thus the implementation of SRT should also be taken into consideration when evaluating the comorbid effects of HT on HCM.
In this study, we investigated 696 HCM patients whose diagnosis of HCM was confirmed by the genetic test and elucidated if the concomitant HT may affect the disease trajectory and morbidity of HCM in the SRT cohort and the non-SRT cohort separately.
METHODS
Study Population
A total of 1395 unrelated patients with HCM were prospectively enrolled at Fuwai Hospital, National Center for Cardiovascular Diseases, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China. All patients underwent a genetic test (panel or whole-exome sequencing) using genomic DNA isolated from blood samples.[10,11] Eight sarcomeric genes (MYH7, MYBPC3, TNNT2, TNNI3, MYL2, MYL3, TPM1, and ACTC1) were evaluated. Variants were classified by pathogenicity as pathogenic (P), likely pathogenic (LP), variants of unknown significance (VUS), likely benign, or benign according to the American College of Medical Genetics and Genomics criteria.
Diagnosis of genetically confirmed HCM was determined by two types of criteria in this study: (1) phenotypic criteria: echocardiographic and/or cardiac magnetic resonance demonstration of LV hypertrophy (maximal LV wall thickness ≥ 15 mm or ≥ 13 mm in patients with a family history of HCM in the absence of any other cardiac or systemic disease capable of producing such a magnitude of hypertrophy;[12] and (2) genotypic criteria: carrying either disease-causing P/LP variants or VUS. Finally, 696 patients with genetically confirmed HCM were included in the analysis.
Informed consent was obtained from all patients in accordance with the principle of the Declaration of Helsinki and the study was approved by the Ethics Committee of Fuwai Hospital (No.2018-989), National Center for Cardiovascular Diseases, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China.
Determination of HCM with HT
HT was diagnosed as blood pressure ≥ 140/90 mmHg in adults or > 95th percentile in adolescents, adjusted for height, age, and sex, which was measured on three separate occasions, or the use of anti-hypertensive drugs.[13] HCM with HT was defined as clinically diagnosed HCM in patients with the following conditions: (1) a history of HT; and (2) diagnosed HT at this admission. All HCM diagnoses and HT diagnoses were made by experienced cardiologists and radiologists.
Measurement of N-terminal Pro-B-type Natriuretic Peptide (NT-proBNP) Levels
NT-proBNP levels were measured in 476 patients (251 patients in the SRT cohort and 225 patients in the non-SRT cohort) at enrollment. The method for measuring NT-proBNP was performed as previously described.[14]
Evaluation of Late Gadolinium Enhancement (LGE)
LGE was evaluated in 293 patients (143 patients in the SRT cohort and 150 patients in the non-SRT cohort) at enrollment as previously described.[15] The extent of LGE (%) was quantitatively assessed using the American Heart Association 17-segment model.[16] With this model, the LV is divided into three sections: base, mid-cavity, and apex; and further subdivided into 17-segments: 6 basal segments (basal anterior, basal anteroseptal, basal inferoseptal, basal inferior, basal inferolateral, and basal anterolateral), 6 mid-cavity segments (mid anterior, mid anteroseptal, mid inferoseptal, mid inferior, mid inferolateral, and mid anterolateral), 4 apical segments (apical anterior, apical septal, apical inferior, and apical lateral), and the true apex as segment 17. It needs to be noted that the 17th segment (i.e., apical cap) could not be identified by the short axis scan,[17] thus the apical cap is not separately assessed on our cardiovascular magnetic resonance myocardial imaging.
Follow-up and Outcomes
All patients were followed up annually until July 2020, and outcome data were obtained by telephone interview, follow-up letter, or clinic visit. All events were carefully recorded, checked, and verified by an independent group of clinical physicians. Investigator training and blinded questionnaires were performed to obtain high-quality data. The primary endpoint was cardiovascular death. The secondary endpoint was all-cause death. Cardiovascular death was considered as sudden cardiac death, heart failure-related death, or stroke-related death.
Statistical Analysis
Statistical analysis was performed using SPSS 26.0 (SPSS Inc., IBM, Armonk, NY, USA) and R statistical software 4.1.1 (http://www.r-proje ct.org). Continuous data were expressed as the mean ± SD or the median [interquartile range (IQR)]. Categorical variables were summarized as counts (percentages). The independent Student’s t-test was used to compare continuous variables between groups and the Pearson’s chi-squared test was used to compare categorical variables. Two-sided P-value < 0.05 were considered statistically significant.
The inverse probability of treatment weighting (IPTW) based on propensity score was used to reduce the potential bias. The propensity scores were generated by a multivariable logistic regression model. The weights of individual patients were estimated by propensity scores.[18] In the primary analysis, all baseline variables including demographics (age and gender), clinical features [body mass index (BMI), atrial fibrillation, syncope, non-sustained ventricular tachycardia, family history, duration of HCM before enrollment], echocardiographic variables [maximal LV wall thickness, LV end-diastolic diameter (LVEDD), LV ejection fraction, left atrial diameter and LV outflow tract obstruction (defined as LV outflow tract gradient ≥ 30 mmHg)], and genetic characteristics were adjusted by IPTW for outcome analyses. The outcome analyses were also performed in patients with both available NT-proBNP and LGE data as a complete data analysis. NT-proBNP levels and the extent of LGE, along with the other variables mentioned above, were enrolled in the IPTW adjustment in the complete data analysis.
Cox proportional hazards regression was performed to calculate the hazard ratio (HR) with 95% confidence interval (CI) and to evaluate the associations between HT and outcomes in HCM. The variables that could not be balanced by IPTW were further incorporated into the Cox model for adjustment.
RESULTS
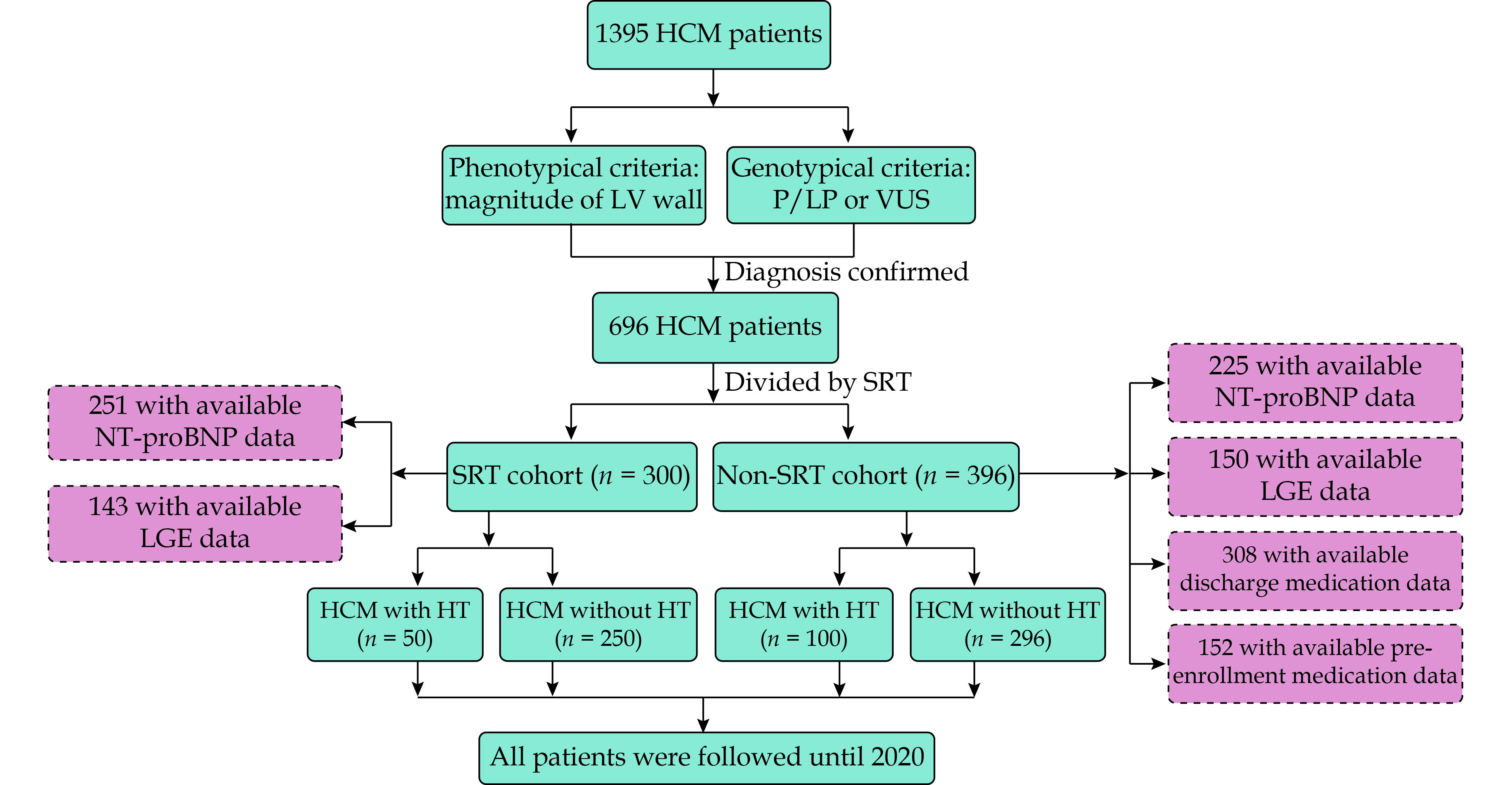
A total of 696 patients with genetically confirmed HCM were enrolled in this study, which consisted of 300 patients (43.1%) who underwent SRT and 396 patients (56.9%) who did not. The flow chart of this study was shown in Figure 1.
Large Pathophysiological and Prognostic Heterogeneity between Patients with and without SRT
As shown in supplemental material, Table 1S, 65 of 300 HCM patients (21.7%) who underwent SRT experienced syncope, the percentage of which was much higher than patients free from SRT (9.3%, P < 0.001). Meanwhile, patients who underwent SRT had a significantly thicker LV wall (25.1 ± 5.4 mm vs. 22.4 ± 5.3 mm, P < 0.001), a larger left atrium (43.4 ± 7.1 mm vs. 41.1 ± 7.8 mm, P < 0.001) and an overwhelming LV outflow tract obstruction (87.7% vs. 27.0%, P < 0.001) compared with those free from SRT. The outcome analyses clearly showed that HCM patients who underwent SRT had a significantly better prognosis, including cardiovascular death (IPTW-adjusted HR = 0.300, 95% CI: 0.140–0.641, P = 0.002) and all-cause death (IPTW-adjusted HR = 0.345, 95% CI: 0.152–0.780, P = 0.011) compared with those who did not undergo SRT (supplemental material, Table 2S). These results indicated the large pathophysiological and prognostic heterogeneity between HCM patients who underwent SRT and patients who did not. Therefore, we conducted a stratified analysis to elucidate the actual differences in clinical characteristics and outcomes between HCM patients with and without HT in the SRT cohort and the non-SRT cohort separately.
Clinical Characteristics and Outcomes of HCM Patients with and without HT in the SRT Cohort
The SRT cohort (n = 300) was composed of 50 HCM patients with HT and 250 HCM patients without HT. Compared with patients with HT, patients without HT were younger (39.4 ± 14.7 years vs. 51.1 ± 8.3 years, P < 0.001). Moreover, compared with patients with HT, BMI was lower (24.2 ± 3.3 kg/m2vs. 26.2 ± 3.5 kg/m2, P < 0.001), family history of HCM (33.6% vs. 16.0%, P = 0.014) was more frequent, and LVEDD was smaller (41.2 ± 5.3 mm vs. 43.4 ± 4.9 mm, P = 0.007) in patients without HT (Table 1).
Table
1.
The baseline and genetic characteristics of genetically confirmed HCM patients with and without HT in the SRT cohort and the non-SRT cohort.
SRT cohort
Non-SRT cohort
Original cohort
Weighted cohort
Original cohort
Weighted cohort
HCM without HT (n = 250)
HCM with HT (n = 50)
P-value
HCM without HT
HCM with HT
P-value
HCM without HT (n = 296)
HCM with HT (n = 100)
P-value
HCM without HT
HCM with HT
P-value
Age, yrs
39.4 ± 14.7
51.1 ± 8.3
< 0.001
41.4 ± 14.7
47.4 ± 7.6
< 0.001
43.5 ± 14.4
57.3 ± 12.0
< 0.001
47.6 ± 15.2
52.0 ± 13.4
0.048
Male
172 (68.8%)
34 (68.0%)
0.911
69.2%
74.6%
0.549
186 (62.8%)
61 (61.0%)
0.743
59.7%
63.0%
0.640
Clinical features
Body mass index, kg/m2
24.2 ± 3.3
26.2 ± 3.5
0.001
24.6 ± 3.3
25.3 ± 3.7
0.337
24.8 ± 3.5
26.5 ± 3.4
< 0.001
25.1 ± 3.7
25.9 ± 3.3
0.102
Atrial fibrillation
36 (14.4%)
12 (24.0%)
0.091
16.4%
21.2%
0.536
63 (21.3%)
23 (23.0%)
0.719
20.2%
23.5%
0.564
Syncope
58 (23.2%)
7 (14.0%)
0.149
21.5%
20.7%
0.939
29 (9.8%)
8 (8.0%)
0.593
8.7%
6.1%
0.400
Non-sustained ventricular tachycardia
14 (5.6%)
1 (2.0%)
0.477
5.0%
2.0%
0.360
20 (6.8%)
5 (5.0%)
0.532
5.9%
5.9%
0.988
Family history of HCM
84 (33.6%)
8 (16.0%)
0.014
30.9%
23.6%
0.460
121 (40.9%)
20 (20.0%)
< 0.001
34.0%
26.7%
0.317
Family history of sudden cardiac death
42 (16.8%)
4 (8.0%)
0.115
15.3%
11.4%
0.649
67 (22.6%)
15 (15.0%)
0.103
21.2%
18.9%
0.723
Pacemakers
20 (8.0%)
3 (6.0%)
0.846
8.0%
16.2%
0.292
28 (9.5%)
5 (5.0%)
0.236
8.0%
6.9%
0.791
Implantable cardioverter defibrillator
0
0
NA
0
0
NA
5 (1.7%)
1 (1.0%)
0.989
1.5%
1.0%
0.719
Duration of HCM before enrollment, yrs
4.0 (1.0–6.8)*
4.0 (1.0–6.0)*
0.765
4.0 (1.0–6.0)*
4.0 (0.0–6.0)*
0.710
4.0 (0.0–10.0)*
4.0 (1.0–10.0)*
0.567
4.0 (0.0–10.0)*
2.9 (1.0–9.9)*
0.853
Echocardiography
Maximal left ventricular wall thickness, mm
25.3 ± 5.4
24.1 ± 5.8
0.190
25.0 ± 5.3
25.2 ± 6.3
0.908
22.9 ± 5.4
21.0 ± 4.7
0.002
22.3 ± 5.2
22.2 ± 5.4
0.932
Left ventricular end-diastolic diameter, mm
41.2 ± 5.3
43.4 ± 4.9
0.007
41.5 ± 5.2
42.3 ± 4.7
0.447
43.6 ± 6.3
45.9 ± 5.6
0.001
44.0 ± 6.0
44.8 ± 5.7
0.311
Left ventricular end-diastolic volume index, mL/m2
74.3 ± 14.4
72.5 ± 15.9
0.412
74.1 ± 14.0
73.5 ± 15.3
0.807
69.5 ± 10.2
68.3 ± 11.1
0.333
69.1 ± 10.5
68.0 ± 10.3
0.429
Left ventricular ejection fraction, %
70.1 ± 7.1
70.6 ± 6.3
0.621
70.1 ± 7.0
70.7 ± 5.7
0.571
65.9 ± 8.8
65.2 ± 7.5
0.497
65.7 ± 8.9
65.9 ± 7.7
0.869
Left atrial diameter, mm
43.1 ± 7.0
44.8 ± 7.9
0.127
43.5 ± 7.0
45.3 ± 8.0
0.230
40.7 ± 7.9
42.2 ± 7.2
0.083
41.0 ± 7.6
42.0 ± 6.3
0.232
Left ventricular outflow tract obstruction
219 (87.6%)
44 (88.0%)
0.937
87.4%
81.5%
0.427
80 (27.0%)
27 (27.0%)
0.996
26.2%
29.1%
0.666
Variants identified
MYH7
140 (56.0%)
23 (46.0%)
0.195
54.2%
45.5%
0.396
136 (45.9%)
36 (36.0%)
0.056
42.5%
36.6%
0.407
MYBPC3
91 (36.4%)
17 (34.0%)
0.747
36.4%
47.3%
0.293
119 (40.2%)
47 (47.0%)
0.234
41.0%
47.5%
0.371
TNNT2
13 (5.2%)
4 (8.0%)
0.655
5.7%
4.7%
0.750
23 (7.8%)
7 (7.0%)
0.801
8.3%
7.4%
0.830
TNNI3
7 (2.8%)
4 (8.0%)
0.169
3.7%
3.4%
0.894
22 (7.4%)
8 (8.0%)
0.853
7.1%
7.5%
0.920
TPM1
6 (2.4%)
1 (2.0%)
1.000
2.3%
1.4%
0.649
6 (2.0%)
3 (3.0%)
0.860
1.8%
1.5%
0.796
MYL2
11 (4.4%)
1 (2.0%)
0.693
4.0%
1.9%
0.455
10 (3.4%)
3 (3.0%)
1.000
2.9%
2.0%
0.606
MYL3
4 (1.6%)
3 (6.0%)
0.171
2.1%
2.0%
0.966
4 (1.4%)
3 (3.0%)
0.520
4.1%
1.9%
0.429
ACTC1
2 (0.8%)
0
1.000
0.7%
0
0.217
3 (1.0%)
1 (1.0%)
1.000
1.0%
1.0%
0.990
Data are presented as means ± SD or n (%). *Presented as median (interquartile range). HCM: hypertrophic cardiomyopathy; HT: hypertension; NA: not available; SRT: septal reduction therapy.
NT-proBNP levels were measured in 251 patients and were found to be comparable between patients without and with HT [1410.5 (753.1–2453.0) ng/L vs. 1087.0 (481.9–2389.6) ng/L, P = 0.172] (supplemental material, Table 3S). Moreover, 143 patients underwent LGE evaluation, patients without HT showed a similar extent of LGE to those with HT (Figure 2 and supplemental material, Table 4S).
Figure
2.
Visualization of the segmental extent of LGE in HCM.
(A): Schematic diagram of the 16-segment heart model; (B): the segmental extent of LGE in HCM without HT patients in the SRT cohort; (C): the segmental extent of LGE in HCM with HT patients in the SRT cohort; (D): the segmental extent of LGE in HCM without HT patients in the non-SRT cohort; and (E): the segmental extent of LGE in HCM with HT patients in the non-SRT cohort. HCM: hypertrophic cardiomyopathy; HT: hypertension; LGE: late gadolinium enhancement; SRT: septal reduction therapy.
The constituent ratio of eight disease-causing sarcomeric gene variants (including P/LP variants and VUS) was found to be comparable between patients with and without HT in the SRT cohort (Table 1).
The median follow-up time was 4.9 years in the SRT cohort. Cardiovascular death and all-cause death occurred in 13 patients (4.3%) and 15 patients (5.0%), respectively. In the primary analyses, baseline characteristics were well balanced after weighting, except for age (Table 1); thus, age was further adjusted in the Cox model for estimating HR with 95% CI. Cox proportional hazards regression analyses showed that the risks of cardiovascular death (IPTW-adjusted HR = 4.372, 95% CI: 0.648–29.507, P = 0.130) and all-cause death (IPTW-adjusted HR = 3.175, 95% CI: 0.773–13.029, P = 0.109) were similar between HCM patients with and without HT (Table 2). The complete data analyses were consistent with the outcome results of the primary results (Table 2).
Table
2.
Cox regression analyses of the association between HT and outcomes in genetically confirmed HCM patients.
SRT cohort (IPTW-adjusted)***
Non-SRT cohort (IPTW-adjusted)***
HR (95% CI)
P-value
HR (95% CI)
P-value
Primary analyses
Cardiovascular death
HCM with HT
Reference
Reference
Reference
Reference
HCM without HT
4.372 (0.648–29.507)
0.130
2.537 (1.085–5.932)
0.032
All-cause death
HCM with HT
Reference
Reference
Reference
Reference
HCM without HT
3.175 (0.773–13.029)
0.109
3.309 (1.111–9.850)
0.032
Complete data analyses*
Cardiovascular death
HCM with HT
Reference
Reference
NA
NA
HCM without HT
0.239 (0.021–2.792)
0.254
All-cause death
HCM with HT
Reference
Reference
Reference
Reference
HCM without HT
0.239 (0.021–2.792)
0.254
10.311 (1.088–97.686)
0.042
Pre-match sensitivity analysis**
Cardiovascular death
HCM with HT
NA
NA
Reference
Reference
HCM without HT
4.543 (1.363–15.145)
0.014
All-cause death
HCM with HT
NA
NA
Reference
Reference
HCM without HT
3.995 (1.541–10.349)
0.004
*Referred to analyses in patients with both available N-terminal pro-B-type natriuretic peptide and late gadolinium enhancement data. **Referred to the non-SRT cohort was pre-matched with the same sample size (n = 300) as the SRT cohort. ***Referred to all variables were adjusted by IPTW and variables that could not be balanced were further adjusted in the Cox model. CI: confidence interval; HCM: hypertrophic cardiomyopathy; HR: hazard ratio; HT: hypertension; IPTW: inverse probability of treatment weighting; NA: not available; SRT: septal reduction therapy.
Clinical Characteristics and Outcomes of HCM Patients with and without HT in the Non-SRT Cohort
The non-SRT cohort (n = 396) contained 100 HCM patients with HT and 296 HCM patients without HT. Patients without HT were younger (43.5 ± 14.4 years vs. 57.3 ± 12.0 years, P < 0.001), had a lower BMI (24.8 ± 3.5 kg/m2vs. 26.5 ± 3.4 kg/m2, P < 0.001), a higher frequency of a family history of HCM (40.9% vs. 20.0%, P < 0.001), as well as a smaller LVEDD (43.6 ± 6.3 mm vs. 45.9 ± 5.6 mm, P = 0.007) and a thicker LV wall (22.9 ± 5.4 mm vs. 21.0 ± 4.7 mm, P = 0.002) compared with those with HT (Table 1).
In 225 patients that underwent NT-proBNP measurements, a higher level of NT-proBNP was found in patients without HT than those with HT [1178.3 (641.0–2005.0) ng/L vs. 763.9 (386.8–1457.8) ng/L, P = 0.004] (supplemental material, Table 3S). The extent of LGE was evaluated in 150 patients, and results showed that patients without HT presented with a higher extent of LV global LGE than those with HT [6.7% (3.1%–14.0%) vs. 4.8% (1.7%–8.3%), P = 0.028]. In the more detailed regional analyses, a higher extent of LGE (patients without HT vs. patients with HT) in midventricular [7.6% (3.0%–16.2%) vs. 3.7% (1.6%–9.8%), P = 0.027], apical [2.7% (0.7%–8.0%) vs. 1.6% (0.2%–4.0%), P = 0.029], septum [7.4% (3.8%–17.8%) vs. 5.0% (1.9%–9.6%), P = 0.012], and inferior [4.4% (0.9%–10.5%) vs. 2.7% (0.3%–5.3%), P = 0.044] areas was indicated in patients without HT (supplemental material, Table 4S). The distribution of LGE was inhomogeneous and asymmetric in patients with and without HT (Figure 2).
There were no significant differences in the constituent ratio of eight disease-causing sarcomeric gene variants between patients with and without HT in the non-SRT cohort (Table 1).
The median follow-up time was 5.9 years in the non-SRT cohort. Cardiovascular death and all-cause death occurred in 44 patients (11.1%) and 56 patients (14.1%), respectively. In the primary analyses, baseline characteristics were well balanced after weighting, except for age; thus, age was further adjusted in the Cox model for estimating HR with 95% CI. Cox proportional hazards regression analyses showed that the risks of cardiovascular death (IPTW-adjusted HR = 2.537, 95% CI: 1.085–5.932, P = 0.032) and all-cause death (IPTW-adjusted HR = 3.309, 95% CI: 1.111–9.850, P = 0.032) were significantly higher in patients without HT than those with HT (Table 2). In the complete data analyses, all cases of cardiovascular death occurred in patients without HT, and these patients also showed a higher risk of all-cause death (IPTW-adjusted HR = 10.311, 95% CI: 1.088–97.686, P = 0.042) (Table 2).
To ensure that our findings were not affected by the different sample sizes of the two cohorts, we assembled a pre-match non-SRT cohort with the same sample size (n = 300) as the SRT cohort by propensity score matching as a sensitivity analysis. The baseline characteristics of the pre-match non-SRT cohort were summarized (supplemental material, Table 5S). Subsequent outcome analyses showed that the associations between HT and both endpoints remained significant (cardiovascular death: IPTW-adjusted HR = 4.543, 95% CI: 1.363–15.145, P = 0.014; all-cause death: IPTW-adjusted HR = 3.995, 95% CI: 1.541–10.349, P = 0.004) (Table 2) in the pre-match non-SRT cohort, suggesting the robustness of our findings.
Post-hoc Analyses in the Non-SRT Cohort
To determine the factors accounting for the worse conditions in HCM patients without HT compared to patients with HT, we conducted post-hoc analyses on the pre-enrollment and discharge medications used in the non-SRT cohort. The data on pre-enrollment and discharge medications were collected from case records, and were available for 152 and 308 of the total 396 patients, respectively.
The pre-enrollment medication results showed that patients without HT took fewer medications, including non-dihydropyridine calcium channel blockers (CCBs) (patients without HT vs. patients with HT: 11.2% vs. 24.1%, P = 0.038; containing verapamil and diltiazem) and angiotensin-converting enzyme inhibitors (ACEIs)/angiotensin receptor blockers (ARBs) (5.1% vs. 33.3%, P < 0.001) than those with HT (Table 3).
Table
3.
Post-hoc analysis of patients with available medication data in the non-SRT cohort.
Data are presented as n (%). *Referred to include verapamil and diltiazem. HCM: hypertrophic cardiomyopathy; HT: hypertension; SRT: septal reduction therapy.
Results regarding discharge medications showed that compared with patients with HT, non-dihydropyridine CCBs (patients without HT vs. patients with HT: 11.1% vs. 20.7%, P = 0.027; containing verapamil and diltiazem) and ACEIs/ARBs (18.1% vs. 53.3%, P < 0.001) were also significantly less prescribed for HCM patients without HT (Table 3).
DISCUSSION
This study represents the first investigation of HT in a large genetically confirmed HCM patient cohort. Based on our results, patients without HT were younger, had a lower BMI, more frequently presented with a family history of HCM, and had a smaller LVEDD than those with HT. We also revealed that patients without HT were presented with a thicker LV wall, a higher level of NT-proBNP, and a higher extent of LV global and regional (midventricular, apical, septum, and inferior areas) LGE in the non-SRT cohort. The presence of HT did not alter the distribution pattern of LGE. Genetic features were similar between patients with and without HT in either cohort. The outcome analyses demonstrated that HCM patients without HT were at higher risks of cardiovascular death and all-cause death in the non-SRT cohort. These associations between HT and endpoints remained significant in the sensitivity analysis. Further post-hoc analyses in the non-SRT cohort found that patients without HT received fewer non-dihydropyridine CCBs and ACEIs/ARBs both before enrollment and at discharge (Figure 3).
Figure
3.
Take home figure.
Genetically confirmed HCM patients without HT had a higher level of NT-proBNP, more severe LGE, and higher risks of cardiovascular death and all-cause death than patients with HT. The presence of HT did not alter the distribution pattern of LGE. Further post-hoc analyses in the non-SRT cohort found that patients without HT received fewer non-dihydropyridine calcium channel blockers and angiotensin-converting enzyme inhibitors/angiotensin receptor blockers both before enrollment and at discharge. HCM: hypertrophic cardiomyopathy; HT: hypertension; LGE: late gadolinium enhancement; NT-proBNP: N-terminal pro-B-type natriuretic peptide; SRT: septal reduction therapy.
To date, only a small number of studies based on small sample sizes, have focused on HT in HCM patients. One early study that enrolled 78 HCM patients showed that only the posterior wall thickness was different between HCM patients with and without HT. However, this study did not provide any follow-up data.[6] Another study, including 262 HCM patients, reported that HT did not significantly worsen the symptoms and prognosis of HCM, but this study did not account for the significant influence of certain factors, such as SRT on prognosis.[5] Most importantly, the diagnosis of HCM with HT in these studies was unclear because they could not entirely exclude hypertrophy caused by HT. In this study, we enrolled a large cohort of 696 genetically confirmed HCM patients with complete follow-up data and conducted analyses in the SRT cohort and the non-SRT cohort separately.
HT is considered causal in inducing cardiac hypertrophy and shares part of the hypertrophy spectrum with HCM.[19,20] Since previous morphologically diagnostic strategies can only provide favoring diagnosis and are unable to accurately identify the cause of cardiac hypertrophy in patients (primary HCM versus isolated hypertensive cardiac hypertrophy), making a certain HCM diagnosis in hypertensive patients is challenging for clinicians.[21] While the genetic test as one kind of etiologic exploration opens new horizons for the novel diagnosis of HCM. The sarcomeric mutations are the predominant underlying cause of primary HCM, representing a fundamentally different pathologic process compared with isolated hypertensive cardiac hypertrophy. A positive genetic test in phenotypically expressed HCM can be confirmatory, and often useful when morphologic features are mild, equivocal, or atypical. Moreover, a sarcomere mutation with a genetic test would provide evidence strongly favoring a clinical diagnosis of HCM and potentially resolve the ambiguous clinical diagnoses. Our study provides data as we identified HCM by a confirmed genetic mutation, rather than morphology alone used in other studies. In addition, the genetic confirmation also provides a purer and more consistent etiologic background to evaluate the effects of concomitant HT on HCM.
In the genetic test, the typical interpretation of a genotype-positive result is a disease-causing P/LP variant. However, with increasing evidence, most VUS are considered causal of HCM.[22,23] Moreover, HCM patients with VUS more closely aligned with P/LP patients regarding prognosis and were shown to have a greater risk of adverse outcomes compared with non-variant carriers.[24] Therefore, to contrast with non-variant carriers, we harbored a variant classified as either P/LP or VUS to confirm the diagnosis of HCM.[25] This is the first study to investigate the different clinical characteristics and outcomes of HCM patients with and without HT in a robust genetic background of HCM.
The results from our study clearly showed that older age and a higher BMI were more common in HCM patients with HT compared to those without HT. These conventional risk factors of HT partially accounted for the comorbidity of HT in this group. Conversely, a family history of HCM and a smaller LVEDD were more frequently identified in HCM patients without HT. These factors are more closely related to the course of HCM. In addition, patients without HT had a thicker LV wall, a higher level of NT-proBNP, and more severe LGE in the non-SRT cohort compared with patients with HT, suggesting that these patients have a more severe cardiac injury, which may lead to poorer prognosis.[14,15] It is worth noting that, as shown in Figure 2, although the extent of LGE was different between patients with and without HT, the distribution pattern was similar, suggesting that the presence of HT did not change the asymmetric burden on the myocardium.
Patients who required SRT always have a highly symptomatic LV obstruction and severe hemodynamic derangements, whose pathophysiology is vastly different from those free from SRT.[2] Meanwhile, the implementation of such an approach could also largely affect prognosis, as shown in both our results and previous investigations.[7–9] To address these large pathophysiological and prognostic heterogeneity, HCM patients in this study were stratified by SRT and analyzed separately. Our results showed that the overall prognosis of HCM patients without HT was poorer than those with HT in the non-SRT cohort, but such prognostic divergence was not observed in the SRT cohort. The discrepancy in the prognostic effects of HT in two cohorts was probably because SRT is directed toward the eradication of the structural abnormalities (hypertrophic septum) and correction of hemodynamic derangements, thus the achievement of good risk control in HCM patients. Patients who undergo such procedures are at an overall lower risk, compared with the patients in the non-SRT cohort, which is representative of the natural course of HCM without a myocardial structural correction in the real world.
However, it was pretty surprising to determine the worse clinical conditions and the higher risk in HCM patients without HT than those with HT, because HT is commonly known as a modifiable risk factor for cardiovascular diseases.[26,27] We next tried to figure out the true reason for explaining this phenomenon. Genetic factors were not responsible for the differences in our results. While, it is worth noting that the medical therapy for HCM and HT are highly coincident, including beta-receptor blockers, non-dihydropyridine CCBs, as well as low-dose diuretics and ACEIs/ARBs.[28,29] We considered that the more active usage of these agents in hypertensive patients might underlie the worse clinical conditions and the higher risk in HCM patients without HT.
In the post-hoc analyses, we reviewed the medications used in the non-SRT cohort and showed that non-dihydropyridine CCBs, as well as ACEIs/ARBs, were significantly less prescribed in patients without HT both at pre-enrollment and at discharge. ACEIs are competitive inhibitors of angiotensin-converting enzyme that prevent the conversion of angiotensin I to angiotensin II;[30] while ARBs bind and inhibit angiotensin II type 1 receptor.[31] These two agents have been shown to improve the natural history of ventricular remodeling by inhibiting the renin-angiotensin-aldosterone system, which plays a central role in the pathophysiology of ventricular remodeling.[32] Non-dihydropyridine CCBs were typically known to inhibit the entry of calcium ions into the slow L-type calcium channels in the myocardium during depolarization and show the ability to prolong LV ventricular filling time.[28] New perspectives that non-dihydropyridine CCBs might attenuate myocardial remodeling have emerged recently, and this protective role might be achieved by inhibiting the calcium-dependent proteases and the transforming growth factor-beta signaling pathway.[33,34] Our results suggested that patients who did not undergo invasive procedures might significantly benefit from these additional non-first-line HCM medications because of their role in dealing with cardiac remodeling and improving functional capacity. It is reasonable to speculate that the baseline and prognostic differences observed in this study might stem from the more active administration of these agents in patients with HT. These results imply that active SRT had a substantial beneficial effect on patients meeting the indications; and more active and comprehensive medications may be beneficial for HCM patients without surgical indications or opportunities.
LIMITATIONS
There are several limitations in our study that should be noted. Firstly, this is a single-center study. Patients enrolled in the study were recruited from a tertiary referral hospital, which might have resulted in selection bias and may undermine the generalizability of our findings. Secondly, the data on left atrial volume index and medication compliance were not available in this study to perform further analysis. Last but not least, an analysis in a larger population, with the design concerning patients’ living environment and lifestyle, is needed to get more universal results.
CONCLUSIONS
This study focused on the different clinical characteristics and outcomes of HCM patients with and without HT under a robust genetic background. Our results demonstrated that, in the non-SRT cohort, genetically confirmed HCM patients without HT had worse clinical conditions and higher risks of cardiovascular death and all-cause death than patients with HT. These differences may result from the different medications before enrollment and at discharge, suggesting that more active and comprehensive medications may be beneficial for HCM patients without surgical indications or opportunities.
ACKNOWLEDGMENTS
This study was supported by the National Natural Science Foundation of China (No.81870286), and the CAM Innovation Fund for Medical Science (2022-I2M-1-005 & 2020-I2M-C&T-A-006). All authors had no conflicts of interest to disclose.
Zhou B, Perel P, Mensah GA, et al. Global epidemiology, health burden and effective interventions for elevated blood pressure and hypertension. Nat Rev Cardiol 2021; 18: 785−802. doi: 10.1038/s41569-021-00559-8
[4]
Yildiz M, Oktay AA, Stewart MH, et al. Left ventricular hypertrophy and hypertension. Prog Cardiovasc Dis 2020; 63: 10−21. doi: 10.1016/j.pcad.2019.11.009
[5]
Luo Q, Chen J, Zhang T, et al. Retrospective analysis of clinical phenotype and prognosis of hypertrophic cardiomyopathy complicated with hypertension. Sci Rep 2020; 10: 349. doi: 10.1038/s41598-019-57230-z
[6]
Karam R, Lever HM, Healy BP. Hypertensive hypertrophic cardiomyopathy or hypertrophic cardiomyopathy with hypertension? A study of 78 patients. J Am Coll Cardiol 1989; 13: 580−584. doi: 10.1016/0735-1097(89)90596-2
[7]
Rastegar H, Boll G, Rowin EJ, et al. Results of surgical septal myectomy for obstructive hypertrophic cardiomyopathy: the Tufts experience. Ann Cardiothorac Surg 2017; 6: 353−363. doi: 10.21037/acs.2017.07.07
[8]
Vanderlaan RD, Woo A, Ralph-Edwards A. Isolated septal myectomy for hypertrophic obstructive cardiomyopathy: an update on the Toronto General Hospital experience. Ann Cardiothorac Surg 2017; 6: 364−368. doi: 10.21037/acs.2017.05.12
[9]
Kim LK, Swaminathan RV, Looser P, et al. Hospital volume outcomes after septal myectomy and alcohol septal ablation for treatment of obstructive hypertrophic cardiomyopathy: US nationwide inpatient database, 2003–2011. JAMA Cardiol 2016; 1: 324−332. doi: 10.1001/jamacardio.2016.0252
[10]
Wu G, Liu L, Zhou Z, et al. East Asian-specific common variant in TNNI3 predisposes to hypertrophic cardiomyopathy. Circulation 2020; 142: 2086−2089. doi: 10.1161/CIRCULATIONAHA.120.050384
[11]
Wang J, Wang Y, Zou Y, et al. Malignant effects of multiple rare variants in sarcomere genes on the prognosis of patients with hypertrophic cardiomyopathy. Eur J Heart Fail 2014; 16: 950−957. doi: 10.1002/ejhf.144
[12]
Ommen SR, Mital S, Burke MA, et al. 2020 AHA/ACC guideline for the diagnosis and treatment of patients with hypertrophic cardiomyopathy: executive summary: a report of the American College of Cardiology/American Heart Association Joint Committee on clinical practice guidelines. Circulation 2020; 142: e533−e557. doi: 10.1161/CIR.0000000000000938
[13]
Wu X, Yuan X, Wang W, et al. Value of a machine learning approach for predicting clinical outcomes in young patients with hypertension. Hypertension 2020; 75: 1271−1278. doi: 10.1161/HYPERTENSIONAHA.119.13404
[14]
Wu G, Liu J, Wang S, et al. N-terminal pro-brain natriuretic peptide and sudden cardiac death in hypertrophic cardiomyopathy. Heart 2021; 107: 1576−1583. doi: 10.1136/heartjnl-2020-317701
[15]
Liu J, Zhao S, Yu S, et al. Patterns of replacement fibrosis in hypertrophic cardiomyopathy. Radiology 2022; 302: 298−306. doi: 10.1148/radiol.2021210914
[16]
Schulz-Menger J, Bluemke DA, Bremerich J, et al. Standardized image interpretation and post-processing in cardiovascular magnetic resonance--2020 update: Society for Cardiovascular Magnetic Resonance (SCMR): board of trustees task force on standardized post-processing. J Cardiovasc Magn Reson 2020; 22: 19. doi: 10.1186/s12968-020-00610-6
[17]
Shaw LJ, Berman DS, Picard MH, et al. Comparative definitions for moderate-severe ischemia in stress nuclear, echocardiography, and magnetic resonance imaging. JACC Cardiovasc Imaging 2014; 7: 593−604. doi: 10.1016/j.jcmg.2013.10.021
[18]
Yang JY, Webster-Clark M, Lund JL, et al. Propensity score methods to control for confounding in observational cohort studies: a statistical primer and application to endoscopy research. Gastrointest Endosc 2019; 90: 360−369. doi: 10.1016/j.gie.2019.04.236
[19]
Loncaric F, Nunno L, Mimbrero M, et al. Basal ventricular septal hypertrophy in systemic hypertension. Am J Cardiol 2020; 125: 1339−1346. doi: 10.1016/j.amjcard.2020.01.045
[20]
Elliott PM, Anastasakis A, Borger MA, et al. 2014 ESC guidelines on diagnosis and management of hypertrophic cardiomyopathy: the task force for the diagnosis and management of hypertrophic cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J 2014; 35: 2733−2779. doi: 10.1093/eurheartj/ehu284
[21]
Kato TS, Noda A, Izawa H, et al. Discrimination of nonobstructive hypertrophic cardiomyopathy from hypertensive left ventricular hypertrophy on the basis of strain rate imaging by tissue Doppler ultrasonography. Circulation 2004; 110: 3808−3814. doi: 10.1161/01.CIR.0000150334.69355.00
[22]
Aronson SJ, Clark EH, Varugheese M, et al. Communicating new knowledge on previously reported genetic variants. Genet Med 2012; 14: 713−719. doi: 10.1038/gim.2012.19
[23]
Walsh R, Thomson KL, Ware JS, et al. Reassessment of Mendelian gene pathogenicity using 7855 cardiomyopathy cases and 60,706 reference samples. Genet Med 2017; 19: 192−203. doi: 10.1038/gim.2016.90
[24]
Ho CY, Day SM, Ashley EA, et al. Genotype and lifetime burden of disease in hypertrophic cardiomyopathy: insights from the Sarcomeric Human Cardiomyopathy Registry (SHaRe). Circulation 2018; 138: 1387−1398. doi: 10.1161/CIRCULATIONAHA.117.033200
[25]
Harper AR, Goel A, Grace C, et al. Common genetic variants and modifiable risk factors underpin hypertrophic cardiomyopathy susceptibility and expressivity. Nat Genet 2021; 53: 135−142. doi: 10.1038/s41588-020-00764-0
[26]
Weber MA. Coronary heart disease and hypertension. Am J Hypertens 1994; 7: 146S−153S. doi: 10.1093/ajh/7.10.146S
[27]
Kokubo Y, Matsumoto C. Hypertension is a risk factor for several types of heart disease: review of prospective studies. Adv Exp Med Biol 2017; 956: 419−426. doi: 10.1007/5584_2016_99
[28]
Ammirati E, Contri R, Coppini R, et al. Pharmacological treatment of hypertrophic cardiomyopathy: current practice and novel perspectives. Eur J Heart Fail 2016; 18: 1106−1118. doi: 10.1002/ejhf.541
[29]
Williams B, Mancia G, Spiering W, et al. 2018 ESC/ESH guidelines for the management of arterial hypertension. Eur Heart J 2018; 39: 3021−3104. doi: 10.1093/eurheartj/ehy339
[30]
Herman LL, Padala SA, Ahmed I, et al. Angiotensin converting enzyme inhibitors (ACEI). In: StatPearls. Treasure Island (FL): StatPearls Publishing; August 5, 2022.
[31]
Hill RD, Vaidya PN. Angiotensin II receptor blockers (ARB). In: StatPearls. Treasure Island (FL): StatPearls Publishing; March 28, 2022.
[32]
Weber KT, Brilla CG, Campbell SE, et al. Myocardial fibrosis: role of angiotensin II and aldosterone. Basic Res Cardiol 1993; 88: S107−S124. doi: 10.1007/978-3-642-72497-8_8
[33]
Ho CY, Lakdawala NK, Cirino AL, et al. Diltiazem treatment for pre-clinical hypertrophic cardiomyopathy sarcomere mutation carriers: a pilot randomized trial to modify disease expression. JACC Heart Fail 2015; 3: 180−188. doi: 10.1016/j.jchf.2014.08.003
[34]
Mendes AS, Blascke de Mello MM, Parente JM, et al. Verapamil decreases calpain-1 and matrix metalloproteinase-2 activities and improves hypertension-induced hypertrophic cardiac remodeling in rats. Life Sci 2020; 244: 117153. doi: 10.1016/j.lfs.2019.117153
Table
1.
The baseline and genetic characteristics of genetically confirmed HCM patients with and without HT in the SRT cohort and the non-SRT cohort.
SRT cohort
Non-SRT cohort
Original cohort
Weighted cohort
Original cohort
Weighted cohort
HCM without HT (n = 250)
HCM with HT (n = 50)
P-value
HCM without HT
HCM with HT
P-value
HCM without HT (n = 296)
HCM with HT (n = 100)
P-value
HCM without HT
HCM with HT
P-value
Age, yrs
39.4 ± 14.7
51.1 ± 8.3
< 0.001
41.4 ± 14.7
47.4 ± 7.6
< 0.001
43.5 ± 14.4
57.3 ± 12.0
< 0.001
47.6 ± 15.2
52.0 ± 13.4
0.048
Male
172 (68.8%)
34 (68.0%)
0.911
69.2%
74.6%
0.549
186 (62.8%)
61 (61.0%)
0.743
59.7%
63.0%
0.640
Clinical features
Body mass index, kg/m2
24.2 ± 3.3
26.2 ± 3.5
0.001
24.6 ± 3.3
25.3 ± 3.7
0.337
24.8 ± 3.5
26.5 ± 3.4
< 0.001
25.1 ± 3.7
25.9 ± 3.3
0.102
Atrial fibrillation
36 (14.4%)
12 (24.0%)
0.091
16.4%
21.2%
0.536
63 (21.3%)
23 (23.0%)
0.719
20.2%
23.5%
0.564
Syncope
58 (23.2%)
7 (14.0%)
0.149
21.5%
20.7%
0.939
29 (9.8%)
8 (8.0%)
0.593
8.7%
6.1%
0.400
Non-sustained ventricular tachycardia
14 (5.6%)
1 (2.0%)
0.477
5.0%
2.0%
0.360
20 (6.8%)
5 (5.0%)
0.532
5.9%
5.9%
0.988
Family history of HCM
84 (33.6%)
8 (16.0%)
0.014
30.9%
23.6%
0.460
121 (40.9%)
20 (20.0%)
< 0.001
34.0%
26.7%
0.317
Family history of sudden cardiac death
42 (16.8%)
4 (8.0%)
0.115
15.3%
11.4%
0.649
67 (22.6%)
15 (15.0%)
0.103
21.2%
18.9%
0.723
Pacemakers
20 (8.0%)
3 (6.0%)
0.846
8.0%
16.2%
0.292
28 (9.5%)
5 (5.0%)
0.236
8.0%
6.9%
0.791
Implantable cardioverter defibrillator
0
0
NA
0
0
NA
5 (1.7%)
1 (1.0%)
0.989
1.5%
1.0%
0.719
Duration of HCM before enrollment, yrs
4.0 (1.0–6.8)*
4.0 (1.0–6.0)*
0.765
4.0 (1.0–6.0)*
4.0 (0.0–6.0)*
0.710
4.0 (0.0–10.0)*
4.0 (1.0–10.0)*
0.567
4.0 (0.0–10.0)*
2.9 (1.0–9.9)*
0.853
Echocardiography
Maximal left ventricular wall thickness, mm
25.3 ± 5.4
24.1 ± 5.8
0.190
25.0 ± 5.3
25.2 ± 6.3
0.908
22.9 ± 5.4
21.0 ± 4.7
0.002
22.3 ± 5.2
22.2 ± 5.4
0.932
Left ventricular end-diastolic diameter, mm
41.2 ± 5.3
43.4 ± 4.9
0.007
41.5 ± 5.2
42.3 ± 4.7
0.447
43.6 ± 6.3
45.9 ± 5.6
0.001
44.0 ± 6.0
44.8 ± 5.7
0.311
Left ventricular end-diastolic volume index, mL/m2
74.3 ± 14.4
72.5 ± 15.9
0.412
74.1 ± 14.0
73.5 ± 15.3
0.807
69.5 ± 10.2
68.3 ± 11.1
0.333
69.1 ± 10.5
68.0 ± 10.3
0.429
Left ventricular ejection fraction, %
70.1 ± 7.1
70.6 ± 6.3
0.621
70.1 ± 7.0
70.7 ± 5.7
0.571
65.9 ± 8.8
65.2 ± 7.5
0.497
65.7 ± 8.9
65.9 ± 7.7
0.869
Left atrial diameter, mm
43.1 ± 7.0
44.8 ± 7.9
0.127
43.5 ± 7.0
45.3 ± 8.0
0.230
40.7 ± 7.9
42.2 ± 7.2
0.083
41.0 ± 7.6
42.0 ± 6.3
0.232
Left ventricular outflow tract obstruction
219 (87.6%)
44 (88.0%)
0.937
87.4%
81.5%
0.427
80 (27.0%)
27 (27.0%)
0.996
26.2%
29.1%
0.666
Variants identified
MYH7
140 (56.0%)
23 (46.0%)
0.195
54.2%
45.5%
0.396
136 (45.9%)
36 (36.0%)
0.056
42.5%
36.6%
0.407
MYBPC3
91 (36.4%)
17 (34.0%)
0.747
36.4%
47.3%
0.293
119 (40.2%)
47 (47.0%)
0.234
41.0%
47.5%
0.371
TNNT2
13 (5.2%)
4 (8.0%)
0.655
5.7%
4.7%
0.750
23 (7.8%)
7 (7.0%)
0.801
8.3%
7.4%
0.830
TNNI3
7 (2.8%)
4 (8.0%)
0.169
3.7%
3.4%
0.894
22 (7.4%)
8 (8.0%)
0.853
7.1%
7.5%
0.920
TPM1
6 (2.4%)
1 (2.0%)
1.000
2.3%
1.4%
0.649
6 (2.0%)
3 (3.0%)
0.860
1.8%
1.5%
0.796
MYL2
11 (4.4%)
1 (2.0%)
0.693
4.0%
1.9%
0.455
10 (3.4%)
3 (3.0%)
1.000
2.9%
2.0%
0.606
MYL3
4 (1.6%)
3 (6.0%)
0.171
2.1%
2.0%
0.966
4 (1.4%)
3 (3.0%)
0.520
4.1%
1.9%
0.429
ACTC1
2 (0.8%)
0
1.000
0.7%
0
0.217
3 (1.0%)
1 (1.0%)
1.000
1.0%
1.0%
0.990
Data are presented as means ± SD or n (%). *Presented as median (interquartile range). HCM: hypertrophic cardiomyopathy; HT: hypertension; NA: not available; SRT: septal reduction therapy.
Table
2.
Cox regression analyses of the association between HT and outcomes in genetically confirmed HCM patients.
SRT cohort (IPTW-adjusted)***
Non-SRT cohort (IPTW-adjusted)***
HR (95% CI)
P-value
HR (95% CI)
P-value
Primary analyses
Cardiovascular death
HCM with HT
Reference
Reference
Reference
Reference
HCM without HT
4.372 (0.648–29.507)
0.130
2.537 (1.085–5.932)
0.032
All-cause death
HCM with HT
Reference
Reference
Reference
Reference
HCM without HT
3.175 (0.773–13.029)
0.109
3.309 (1.111–9.850)
0.032
Complete data analyses*
Cardiovascular death
HCM with HT
Reference
Reference
NA
NA
HCM without HT
0.239 (0.021–2.792)
0.254
All-cause death
HCM with HT
Reference
Reference
Reference
Reference
HCM without HT
0.239 (0.021–2.792)
0.254
10.311 (1.088–97.686)
0.042
Pre-match sensitivity analysis**
Cardiovascular death
HCM with HT
NA
NA
Reference
Reference
HCM without HT
4.543 (1.363–15.145)
0.014
All-cause death
HCM with HT
NA
NA
Reference
Reference
HCM without HT
3.995 (1.541–10.349)
0.004
*Referred to analyses in patients with both available N-terminal pro-B-type natriuretic peptide and late gadolinium enhancement data. **Referred to the non-SRT cohort was pre-matched with the same sample size (n = 300) as the SRT cohort. ***Referred to all variables were adjusted by IPTW and variables that could not be balanced were further adjusted in the Cox model. CI: confidence interval; HCM: hypertrophic cardiomyopathy; HR: hazard ratio; HT: hypertension; IPTW: inverse probability of treatment weighting; NA: not available; SRT: septal reduction therapy.
Data are presented as n (%). *Referred to include verapamil and diltiazem. HCM: hypertrophic cardiomyopathy; HT: hypertension; SRT: septal reduction therapy.


 DownLoad:
DownLoad:


